Science: Chemistry:
Concept of Enzymatic Hydrogen Production by Light-Sensitized Anodized
Tubular TiO2 Photoanode
Dr.
Hyunku Joo and Jin-Wook Ha*
Korea
Institute of Energy Research,
*
Department of Energy and Environmental
ABSTRACT
In this article, different approach using a light-sensitized enzymatic(LSE) system has been proposed. The LSE is a way of producing hydrogen by coupling an inorganic
semiconductor with enzymes in a photoelectrochemical configuration. The system uses the intrinsic proton
reduction ability of hydrogenase enzyme in
tandem with an anodic compartment
where the electron donors like water undergo oxidative reaction on light-sensitized photoanode. Generated electrons are separated and moved
through an external circuit within a solar cell, which is applied for an
external bias, to the cathodic compartment and used for the reduction of proton
into hydrogen on the active sites of enzymes. Oxidized ions move to the
cathodic compartment through an ion bridge like nanofiltration(NF) membrane in
this study.
1.
INTRODUCTION
Environmental
remediation by photocatalysis has been an attractive issue over the last decade, while recently numerous studies have focused on hydrogen evolution from water as a clean energy
resource since the invention of photochemical water splitting over TiO2
electrode in 1972. However, the
photocatalytic process has been simultaneously criticized as being uneconomical
compared to other oxidative treatment systems and up-to-date hydrogen
production systems due to its inherently
low efficiency and limitations resulting from the necessity for a novel material, an appropriate light source and immobilization,
which may increase the
overall energy costs[1]. Therefore, research
attention has focused on developing economically feasible photocatalytic systems with future commercial
applications. Comprehensive
reviews on semiconductor particulate systems[2], on TiO2
photocatalyst[3] and on the material-related issues[4-5] for hydrogen
production were published. Compared with other photocatalysts, TiO2
is much more promising as it is stable, non-corrosive, environmentally
friendly, abundant and low cost ever since Fujishima and Honda claimed a total efficiency of solar energy
conversion to hydrogen of only 0.4% using a TiO2 single crystal
as photoanode and Pt as cathode involving a chemical bias imposed by pH
difference between the electrodes[4-5].
Recently, photoanodes with TiO2-based film have been investigated
using techniques like anodization[6-11] and sputtering[12]. The aforementioned
nanotube-film on titanium foil by anodization has been of our great interest
for a system of light-sensitized enzymatic hydrogen production.
In this article, different approach using a light-sensitized enzymatic(LSE) system has been proposed. The LSE is a way of producing hydrogen by coupling an inorganic
semiconductor with enzymes in a photoelectrochemical configuration. The system uses the intrinsic proton
reduction ability of hydrogenase enzyme in
tandem with an anodic compartment
where the electron donors like water undergo oxidative reaction on light-sensitized photoanode. Generated electrons are separated and moved
through an external circuit within a solar cell, which is applied for an
external bias, to the cathodic compartment and used for the reduction of proton
into hydrogen on the active sites of enzymes. Oxidized ions move to the
cathodic compartment through an ion bridge like nanofiltration(NF) membrane in
this study.
Our present work aims to give optimized system
condition concerning photoanode of the anodized tubular TiO2 film on
titanium substrate and proton-reducing enzymes to produce hydrogen, a
renewable, nonpolluting and portable energy source. The trend of the system on
the light-sensitized enzymatic splitting of water was then investigated.
2.
EXPERIMENTAL
2.1.
Sample preparation
All
chemicals were used without further purification. P25 TiO2(Degussa, FRG) was used as a reference
when methylene blue(MB, 95% Showa
chemical, Japan) was used as the probe compound for measuring photocatalytic activity of anodized tubular TiO2
electrode(ATTE). Titanium
foils(0.25 mm thick, 99.6% purity,
2.2. Apparatus and
Analysis
Experiments were mainly conducted in a
two-compartment(anodic and cathodic elements
connected via a nanofiltration
membrane, NF) reactor, as shown in
Fig. 1. The anodic
compartment had a volume of 80 ml(headspace volume ca. 55 ml) and
was a cylindrical-shaped cell. The
anodic compartment contains an
aqueous solution of 1.0M KOH for water splitting unless otherwise noted.
The cathodic compartment was a water-jacketed, cylindrical-shaped cell(80 ml, headspace volume ca. 55 ml) with
Tris-HCl buffer sealed with a silicone rubber gasket. Prior to the reaction, the mixture was
de-aerated with nitrogen gas for 20 min to remove the oxygen in water and
headspace. Each of two terminals from a solar cell panel(10cm´10cm, crystalline silicon, max. 1.0V) was
attached to the ATTE and the
platinum mesh cathode to apply external bias into the system. The light
source used was a 1000 W xenon
lamp(
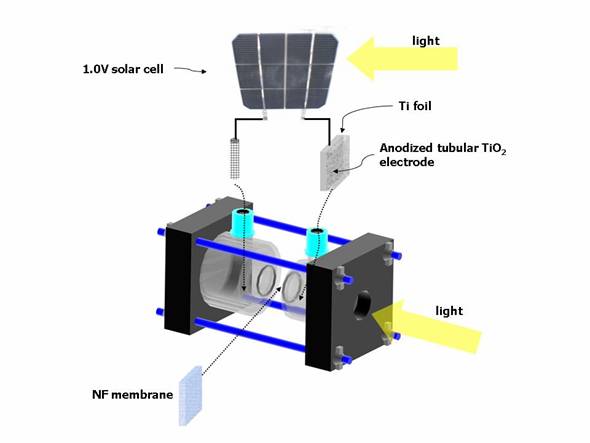
Figure
1. Schematic view of the reactor with anodized tubular TiO2 photoanode,
solar cell
and NF membrane for hydrogen production.
3.
RESULTS AND DISCUSSION
The prepared samples anodized in 0.5M H3PO4+0.14M
NaF+0.1M NaNO3 have tubular(20V of bias, tube length ca. 600 nm) or
web-shaped(30V, thickness ca. 618 nm) TiO2 film on the Ti
substrates. The morphology of porous film was illustrated to change with
electrolytes, concentration of electrolytes, pH of electrolytes, anodizing time
and temperature, and the annealing ambient. This is due to the fact that during
the anodization porous structures are formed through two processes:
field-enhanced oxidation and field-enhanced oxide dissolution. And also, at elevated temperature solid-state sintering is
likely to take place, which leads to grain growth, densification and complete
collapse of the structure that are more remarkable during phase
transformations. The phase transformations often accompany bond-breaking and
enhanced mass transport[6-11]. XRD
analysis of the selected samples indicated
main characteristic anatase peaks
at 2q=25.3 with that of the commercial P25 indicated as a reference when annealed at
lower than 450ºC while rutile
peaks at higher than 500ºC. Ratio of constituents(oxygen
to titanium) of TiO2 film was calculated by EDAX results, which
resulted in the range from 0.1 to 2.4.
Photocatalytic activity of the ATTE
was firstly checked with MB discoloration. The time-coursed profiles of MB discoloration with
the samples along with P25 as a
reference. Unlike P25, the samples revealed lower activity in MB discoloration irrespective of species, while lower dose of P25 showed similar profiles to
the prepared samples. The
Langmuir-Hinshelwood kinetics, generally applied in photocatalysis, indicates
that a relatively high concentration of a probe material converts the equation
to zero order kinetics, evidenced in our study by the simple experiment of
varying the amount of P25 with a
fixed initial concentration of MB(10 ppm) in order to change the time-coursed
profiles of MB discoloration from first order to zero order with decreasing
injection amount. The calculated amount
of TiO2 on Ti substrate(2cm´2cm) turned out 1.6 ~ 3.1mg, assuming that film
thickness is in the range of 1 ~ 2 μm and density of TiO2 is
3.9 g/cm3. This amount is about one tenth of P25(200mg initially).
This is well correlated with the fact that less than 5 mg of P25 showed similar
discoloration rate to the prepared samples.
Secondly, the ATTE itself was irradiated in
0.1M Na2S electrolyte and hydrogen at a rate of ca. 40 μmol/(hr´cm2) was evolved. In this case, Na2S
acted as an electron donor and the ATTE excited the electrons in valence
band(VB) into conduction band(CB), which reduced the protons into hydrogen.
Anodization and annealing condition changed the time profiles of photocatalytic
hydrogen production. Web-shaped TiO2 films were inferior to tubular
TiO2 film such as sample no. 6. It was reported by Khan et al.[13]
that instead of the 3D configuration of particles, fabrication of vertical
standing nano-wires of TiO2 could improve the efficiency. During H2
was produced, the color of the solution in TiO2 film side turned
yellow gradually. This might due to the oxidation of sulfide ion to sulfur(Eo
= -0.47627 vs. SHE). Evacuation of evolved gas in headspace was repeatedly
carried out when the evolution rate decreased, recovering the initial rate.
Hydrogen production or evolution out of water seemed to be dependent of
pressure. Without Na2S a few amount of hydrogen(ca. 0.3 μmol/hr) was produced. When Na2SO3 was added, the solution in
TiO2 film side stayed transparent. In this case, sulfide ion was not
fully oxidized into sulfur, but into S22-(Eo =
-0.42836 vs. SHE). Possible reactions are as follows:
2S2- + 2h+VB → S22- (1)
S22- + SO32- → S2O32- + S2- (2)
S2O32- + 4h+VB + 6OH- → 2SO32- + 3H2O (3)
SO32- + 2h+VB + 2OH- → SO42- + 3H2O (4)
2SO42- + 2h+VB → S2O62- (5)
2H2O + 2e-CB → H2
+
2OH- (6)
Effect of others electrolytes such as 1.0M KOH,
0.1M NaOH, seawater, 0.1M K2SO4 or 1M KCl on the amount
of hydrogen was investigated, resulting in a rate of far less than 1
μmol/(hr´cm2).
Figure 2.
Time-coursed profile of enzymatic
hydrogen evolution with the anodized tubular TiO2
photoanode(annealed at 550ºC, 1.0M
KOH in anodic compartment, Tris-HCl buffer in cathodic compartment, irradiated
photoanode area 1.0 cm2, ca. 68 mW/cm2 irradiation with
Xe lamp).
Thirdly, enzymatic hydrogen
evolution in cathodic compartment with oxygen evolved in the anodic compartment
was conducted with external bias(ca. 1.0V) applied by a solar cell. Previously,
applied potential and its effect was examined with a potentiostat from -1.5V ~
+1.5V. Working electrode(WE) was
placed on either the photoanode(P25 on ITO substrate) or the Pt cathode, while
counter electrode(CE) was on the rest with reference electrode(RE), Ag/AgCl in
saturated KCl. Then, external bias was applied onto the WE by potentiostat from
-1.5 to +1.5V. Positive bias onto the photoanode or negative bias onto the Pt cathode
generated respective negative current or positive current indicating an
electron flow from the photoanode to the cathode for both cases. This upward
shift of the anodic energy level led hydrogen evolution in the cathode
compartment. Otherwise, completely opposite results were obtained without any
hydrogen evolution. Noticeable hydrogen evolution was identified at a photocurrent above
300 μA when 1.3 V bias in absolute value was applied to the electrode. After the aforementioned check-ups, both a solar cell and a photoanode
were irradiated to increase the Fermi level and to excite electrons in the VB
into the CB of the photoanode. This system configuration led to the
stoichiometric evolution of H2 and O2(H2:O2=2:1).
Without any electron donor in the anodic compartment, oxygen was produced at a
rate of ca. 15 μmol/(hr´cm2) and hydrogen was produced in the cathodic compartment at a
rate of ca. 30 μmol/(hr´cm2). It revealed the rate of hydrogen production was ca. 30
μmol/(hr´cm2), which could be translated into ca. 6.75 L/(hr´m2). On a fine sunny day for 6.5 hrs
in the area of 104 m2(100m´100m), 439,000 L of H2 could be
evolved by rough calculation. Assuming that a fuel cell vehicle needs 4.6 kg of
H2 for 395 km, 439,000L of H2 is for more than ten
vehicles to be charged and for 3,352 km in distance.
4.
CONCLUSIONS
For
hydrogen production with solar irradiation in the future, a relatively novel approach was described where sensitized photoanode donates electrons to hydrogenase
that in turn reduces protons into
hydrogen. Vertical standing nanotubes of TiO2 film was stably operating as
a light sensitizer, evolving oxygen and enzymes were efficiently reducing
protons into hydrogen. Practically feasible system configuration was also
investigated replacing a salt bridge with nanofiltration membrane. To increase
the amount of harvested light, longer TiO2 tube with optimum
crystallinity has been under considered.
Acknowledgements
This
research was performed for the
REFERENCES
1. G. B. Raupp, A. Alexiadis, M. Hossain, and R. Changrani, Catalysis Today, 69, 41 (2001).
2. M. Ashokkumar, “An overview on semiconductor
particulate systems for photoreduction of hydrogen”, Int. J. Hydrogen Energy, 23(6), 427-438 (1998).
3. M. Ni, M.K.H. Leung, D.Y.C. Leung, and K.
Sumathy, Renewable and Sustainable Energy
Reviews, 11, 401 (2007).
4. T. Bak, J. Nowotny, M. Rekas, and C. C. Sorrell, Int. J. Hydrogen Energy, 27(1), 19 (2002).
5. T. Bak, J. Nowotny, M. Rekas, and C. C.
Sorrell, Int.
J. Hydrogen Energy, 27(2), 991 (2002).
6. D. Gong, C.A. Grimes, O.K. Varghese, W. Hu,
R.S. Singh, Z. Chen, and E. C. Dickey, “Titanium oxide nanotube arrays prepared
by anodic oxidation”, J. Mater. Res.,
16(12), 3331-3334 (2001).
7. O.K. Varghese, D. Gong, M. Paulose, C.A.
Grimes, and E. C. Dickey, “Crystallization and high-temperature structural
thermal stability of titanium oxide nanotube arrays”, J. Mater. Res., 18(1), 156-165 (2003).
8. G. K. Mor, O. K. Vargheese, M. Paulose, N.
Mukherjee, and C. A. Grimes, J. Mater.
Res. 18, 2588-2591 (2003).
9. G. K. Mor, K. Shankar, M. Paulose, O. K.
Vargheese, and C. A. Grimes, Nanoletters
5, 191-195 (2005).
10. M. Paulose, G. K. Mor, O. K. Vargheese, K.
Shankar, and C. A. Grimes, J. Photochem.
Photobiol. A:Chem. 178, 8-15 (2006).
11. K. S.
Raja, V. K. Mahajan, and M. Misra, J. of
Power Sources, 159, 1258-1265 (2006).
12. M. Kitano, M. Takeuchi, M. Matsuoka, J. M.
Thomas, and M. Anpo, Catalysis Today
120, 133-138 (2007).
13. S.U.M. Khan and T. Sultana, Solar Energy Materials and Solar Cells
76, 211-221 (2003).
[ BWW Society Home Page ]
© 2007 The Bibliotheque: World Wide Society