A
Possible Role of the Glial System in the Pathophysiology
of So-Called Mental Disorders
By Dr. Bernhard J. Mitterauer
1. Introduction
The so-called mental disorders or psychobiological disorders basically
comprise affective psychoses (bipolar disorders or manic-depressive illness)
and delusional disorders like schizophrenia. After the description and
explanation of the pertinent hypotheses, I will deduce the main symptomatology
of the psychiatric disorders from these. Since the theoretical framework proposed
is essentially based on glial-neuronal interactions, let me start out with an
overview of the glial system in its interaction with the neuronal system.
2. Glial neuronal interaction
2.1. General introduction
The nervous tissue of the brain consists of the neuronal system
(neurons, axons, dendrites and the glial system (astrocytes, oligodendrocytes
with myelin sheaths which enfold the axons and microglia). Glial cells outnumber neurons in the central
nervous system (brain) by a factor of 10 to 1 (Kuffler et.al., 1984). Virchow (1846) considered them to be merely
connective tissue between the neurons
(“nerve glue”). Meanwhile, experimental
results are inspiring a major re-examination of the role of glia in the
regulation of neural integration in the central nervous system (Kettenmann and
Ransom, 1995; Haydon, 2001). Glial cells
(particularly astrocytes with their processes that contact or even enfold a
synapse) modulate the “efficacy of synaptic transmission” (Teichberg, 1991;
Mitterauer et. al., 1996; Mitterauer, 2000 b; Oliet et al. 2001). Signals between astrocytes and neurons can be
mediated by glutamate (Gallo and Ghiani,
2000); acetylcholine (Smit et al., 2001) and other neurotransmitters
(Kimelberg, 1998) and by intracellular calcium oscillations in astrocytes which
have been hypothesized to affect synaptic cleft calcium concentrations (Cooper,
1995; Newman and Zahs, 1997) and, subsequently, the amount of neurotransmitter
released from presynaptic terminals. In
addition to modulating synaptic transmission in neuronal cells, astrocytes may
play a direct role in generating pacemaker rhythms (Mitterauer et al, 2000).
According to my hypothesis, glial cells have a spatio-temporal boundary-setting
function in their interaction with the neuronal system in the sense of
information structuring. In other words, glial cells (astrocytes,
oligodendrocytes) divide the brain into spatially limited areas or
compartments, on the one hand, and create functional units in various time
scales with the neurons, on the other hand. At least what the neuronal system
concerns, there is a general consensus in neuroscience that the brain is
compartmentalized (Rall, 1995).
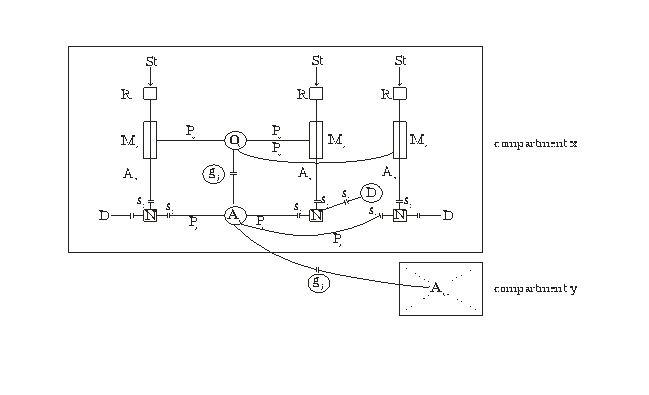 Figure 1. Schematic diagram of two glial-neuronal compartments
Figure 1. Schematic diagram of two glial-neuronal compartments
This figure shows a schematic diagram of two glial-neuronal
compartments. The entire system is composed of the following cell structures:
three receptors (R) are shown that can be occupied by appropriate stimuli (St).
Axons (Ax) lead to the corresponding neurons (N). Three processes (
As I already mentioned, it is experimentally well established that glial
cells exert an active role in modulating the efficacy of synaptic transmission
(Teichberg, 1991; Mitterauer et al, 1996; Oliet et al, 2001; Haydon, 2001;
Mitterauer, 2003).
Now I will mention some experimental indications for the spatial boundary-setting
function of the glial system. Rakic
(1988) proposed an experimentally supported “radial unit hypothesis” for the
development of the cerebral cortex.
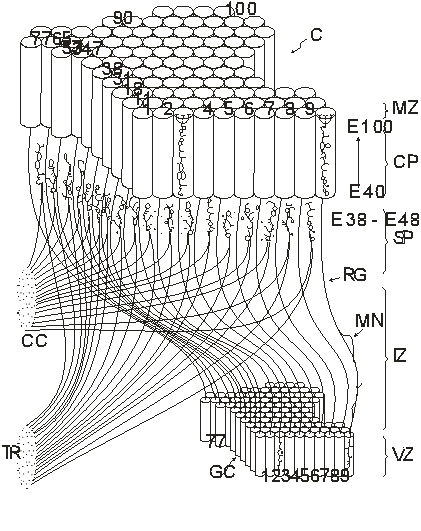
Figure 2. Radial
unit model of the development of the cerebral cortex (Rakic, 1988, with
permission of the author)
This figure shows the relation
between a small patch of the proliferative, ventricular zone (VZ) and its
corresponding area within the cortical plate (CP) in the developing cerebrum.
Although the cerebral surface in primates expands and shifts during prenatal
development, ontogenetic columns (outlined by the cylinders) remain attached to
the corresponding proliferative units by the grid of radial glial fibers (RG). Neurons
produced between E40 (40th embryonic day) and E100 by a given proliferative
unit, migrate in succession along the same radial glial guides (RG) and stack
up in reverse order of arrival within the same ontogenetic column. Each
migrating neuron (MN) first transverses the intermediate zone (IZ) and then the
subplate (SP) which contains interstitial cells and ‘waiting’ afferents from
the thalamic radiation (TR) and ipsilateral and contralateral cortico-cortical
connections (CC) between E38 and E48. After entering the cortical plate, each
neuron bypasses earlier generated neurons and settles at the interface between
the CP and the marginal zone (MZ). As a result, proliferative units 1-100
produce ontogenetic columns 1-100, in the same relative position to each other
without a lateral mismatch… Thus, the specification of cytoarchitectonic areas
and topographic maps depend on the spatial distribution of their ancestors in
the proliferative units, whereas the laminar position and phenotype of neurons
within ontogenetic columns depends on the time of their origin.
According to this hypothesis, the ependymal layer of the embryonic
cerebral ventricle consists of proliferative units that provide a protomap of
prospective cytoarchitectonic areas. The output of the proliferative units is
translated via glial guides to the expanding cortex in the form of ontogenetic
columns, whose final number for each area can be modified through interaction
with afferent input. The radial unit model provides a framework for
understanding cerebral evolution, epigenetic regulation of the parcellation of
cytoarchitectonic areas. According to Rakic, the cerebral cortex develops from
a glial protomap such that an isomorphism exists between the places on the
protomap and the cortical columns. One can also say that radial glial cells
determine the spatial distribution of neurons in the developing cerebral cortex
in the sense of a spatial boundary-setting function. In other words: the radial
glia determine the places where the neurons are at work. Hatten (1990) speaks
of a glial “scaffold”.
This spatial boundary-setting function of the glial system is also
exhibited in the retina, for example, where Müller cells (radial glia) group
neurons into columns. Further, it is established in the olfactory system that
the astroglia have a boundary-setting function in the formation of the
olfactory glomeruli.
2.2. The concept of tripartite synapses
Is there also experimental evidence for the glial temporal
boundary-setting function? An impressive
example of the glial temporal boundary-setting is represented by the model of
tripartite synapses. According to the prevailing view, chemical synaptic
transmission exclusively involves bipartite synapses consisting of presynaptic
and postsynaptic components and a synaptic cleft, in which a presynaptically
released neurotransmitter binds to cognate receptors in the postsynaptic cell.
However, there is a new wave of information suggesting that glia, especially
astrocytes, are intimately involved in the active control of neuronal activity
and synaptic transmission.
Smit and coworkers (2001) proposed a model of a cholinergic tripartite
synapse that might turn out to be a milestone for our understanding of the
glial-neuronal interaction. But first let me shortly describe this type of
tripartite synapse. These authors identified a glia-derived soluble
acetylcholine-binding protein (AChBP), which is a naturally occurring analogue
of the ligand-binding domains of the nicotinic acetylcholine receptors
(nAChRs). Like the nAChRs, it assembles into a heptamer with ligand-binding
characteristics typical of a nicotinic receptor. Presynaptic releases of
acetylcholine induce the secretion of AChBP through the glial secretory
pathway, and once in the synaptic cleft, it acts as a molecular decoy, binding
the transmitter and reducing its availability at the synapse in the sense of a
negative feedback on synaptic efficacy.
This model, which focuses on the role of AChBP in neurotransmission,
suggests that there is a basal level of AChBP in the synaptic cleft, maintained
by continuous release from the synaptic glial cells. Under conditions of active
presynaptic transmitter release, high millimolar concentrations of free ACh
will probably activate both postsynaptic receptors and nAChRs on the synaptic
glial cells, which would enhance the release of AChBP, thus increasing its
concentration in the synaptic cleft. This may either diminish or terminate the
ongoing ACh response or raise the concentration of basal AChBP to the extent
that subsequent responses to ACh are decreased.
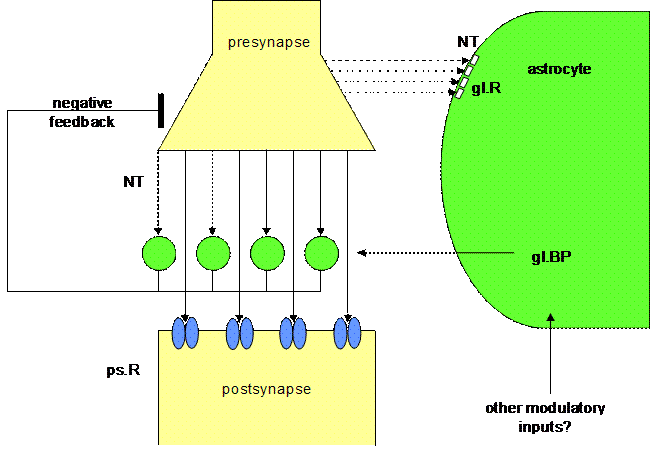
Figure 3. Schematic
diagram of a tripartite synapse
This figure shows a schematic
drawing of a tripartite synapse as proposed by Smit and co-workers (2001), but
generalized for all neurotransmitters (NT). For the sake of clarity, reference
is not made to other modulatory substances such as the functional significance
of calcium waves (Charles and Giaume, 2002; Rose et al, 2003). A
neurotransmitter is released from the presynaptic terminal ready for occupancy
of glial BP and postsynaptic receptors. In parallel, glial receptors are
occupied by neurotransmitters, which increase the production and secretion of
soluble glial BP into the synapse. The increased levels of soluble BP in the
synapse reduces that amount of free neurotransmitter that can bind to
postsynaptic receptors, and neurotransmission is inactivated by this form of
negative feedback. Once the NT levels have returned to baseline, the BP levels
will drop because the glial cells are no longer being stimulated to produce BP;
the synapse will return to its initial state, and synaptic information processing
can start again. In particular, the role of the glutamatergic tripartite
synapse has been documented over the last years (Auld and Robitaille, 2003).
The astrocytes which play a role in these synapses do not make use of a BP, but
produce glutamate as a transmitter by which they negatively feedback
on the presynaptic element of the synapse. In other words, in tripartite
synapses glia have a temporal boundary-setting function in temporarily turning
off synaptic information transmission.
In addition, neuronal synchrony is mediated by astrocytic glutamate
through activation of extrasynaptic N-methyl-D-asparate receptors (Fellin et al, 2004). This is an
experimental verification of our “neuro-glial synchronization hypothesis” (Mitterauer
et al, 1996) in the sense that glia may actively determine temporal processes
in neuronal networks.
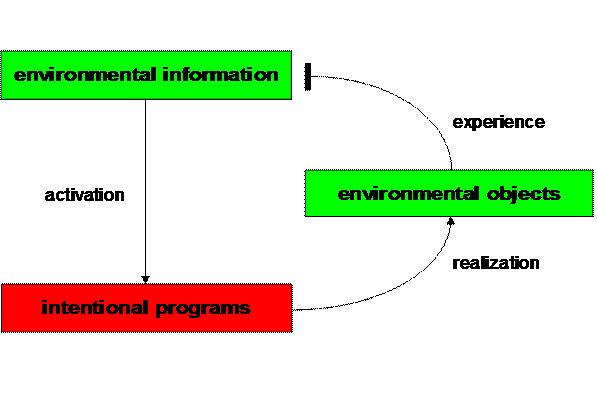
Figure 4. Elementary
behavioral cycle (Mitterauer, 2000)
From a cybernetic point of view, this model of a tripartite synapse can
be described as an elementary behavioral cycle (Mitterauer 2000a; 2004). Such
an interdisciplinary approach could be helpful for interpreting the
pathophysiology of both bipolar disorder and schizophrenia. Generally, a living
system like man is endowed with intentional programs (hungers, desires, etc.)
that strive for realization in the environment (Iberall and McCulloch 1969).
3. Pathophysiological model of so called mental disorders
3.1. Biocybernetic model
A behavioral cycle represents an intentional
relationship of a living system with its environment. Information from the
environment actualizes an intentional program. If a living system is able to
find appropriate objects for realizing a specific intentional program in its
environment, then the cycle is closed and is comparable to an experience based
on a negative feedback mechanism that attenuates the initial positive signal,
thus down regulating information processing (Fig. 4).
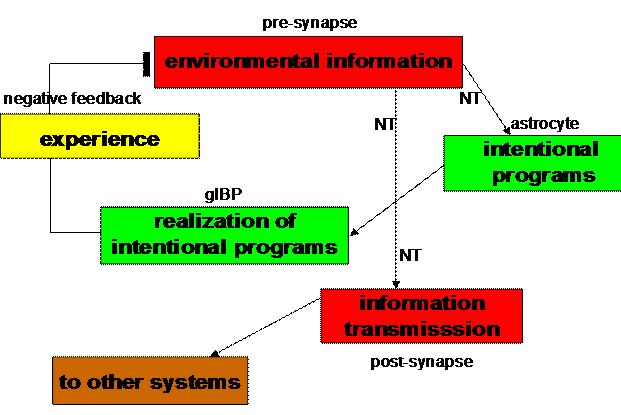
Figure 5. Biocybernetic
model of a tripartite synapse (Mitterauer, 2004)
Such elementary behavioral cycles may also control the information
processing in tripartite synapses. The production of a neurotransmitter (NT) in
the presynapse can be interpreted as “environmental information” stimulating
the expression of glial BP in astrocytes. Glial BP may embody an “intentional
program” that is ready for occupancy by an appropriate neurotransmitter. If an
appropriate occupancy occurs (“realization of the intentional program”), the
glial system negatively feeds back this “experience” to the presynapse. In
parallel, this experience is transmitted to other cells in the glial-neuronal
networks by occupancy of postsynaptic receptors (“information transmission”).
Now, the cycle can start again (Fig. 5).

Figure 6. Balance,
imbalance, and unbalance between neurotransmitter (NT) and its
glial binding protein (gl.BP) (Mitterauer, 2005)
The interaction between
neurotransmitters and glial BP can be system theoretically interpreted (Günther
1963) as balanced, underbalanced, overbalanced, or even unbalanced. Formally
speaking, if the variables (BP) dominate the values (NT) available in the
system, the system is underbalanced. This may be the case in depression. In
contrast, if the values (NT) dominate the variables (BP), the system is
overbalanced, which may occur in manic states (Mitterauer 2004). If no
appropriate variables (gl.BP) are available, the system is totally unbalanced.
Such a synaptic state may be responsible for the pathophysiology of
schizophrenia (Fig. 6).
Now, let me consider in detail the imbalances in tripartite synapses
that may cause the so called mental disorders. How could underbalanced
tripartite synapses cause depression? Can the main symptoms of depression be
explained as a disorder of glial-neuronal interaction in tripartite synapses of
the various neurotransmitter types (Fig. 7)?
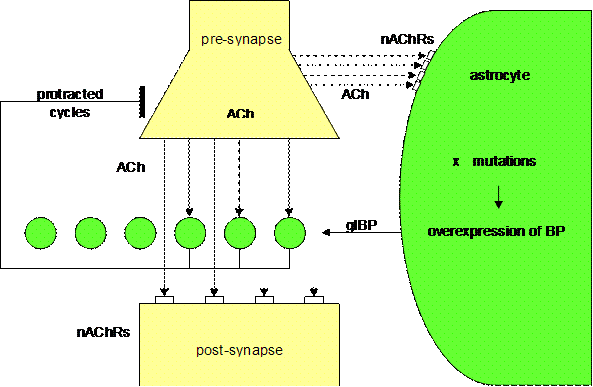
Figure 7. Underbalanced
cholinergic synapse (Mitterauer, 2004)
3.2. Depression
Supposing that genes responsible for the
expression of glial BP are spontaneously or exogenously (stress, etc.) mutated
causing overexpression of glial BP, then the overexpression of glial BP will
reduce the ACh level in the synapse. This relative lack of ACh will result in
an underactivation of both glial nAChRs and postsynaptic receptors as well. The
imbalance of the synaptic glial-neuronal interaction may delay the information
processing in the sense of protracted “behavioral” cycles. This kind of
pathophysiology could be responsible for the psychomotoric retardation of
depressive patients, depending on the transmitter systems or brain areas
affected.
As already proposed, glial BP could embody intentional programs that strive
for realization by means of neurotransmitter occupancy. Hence, an underbalanced
tripartite synapse can be characterized as hyperintentional. Here I see a possible pathophysiological explanation of high
aspirations as typical of depressive patients (Bibring 1953). Considering the
delayed and imperfect information processing in underbalanced tripartite
synapses, feelings of insufficiency may arise on the behavioral level.
Furthermore, the synaptic underbalance could be responsible for the disturbance
of circadian rhythms, a core symptom of depression. Specifically, disturbances
in the synaptic cycle between glial “intentional” programs, presynaptic
“environmental” information and the “experience” of appropriate receptor
occupancy result in a dysfunction of temporal boundary-setting feedback
mechanisms.
The individual and manifold symptomatology of depression may depend on
the types of neurotransmitters or brain regions involved. Since most of the
effective treatments of mood disorder were discovered by empiricism, the
effectiveness of somatic treatment has propelled neurotransmitter theories
rather than vice versa (Post 1995). My approach is contrary to this trend by
deducing the pathophysiology of bipolar disorder from a theoretical model.
Supposing that a lack of neurotransmitter in the synaptic cleft is responsible
for depressive mood, it is conceivable that a treatment with re-uptake
inhibitory substances is successful. At first glance, such a therapeutic
mechanism should also be effective in tripartite synapses, since an increase of
transmitter in the synaptic cleft should balance the excess of glial BP. But
what may occur on the glial nAChRs as described in the model of Smit and
coworkers? The increased concentration of neurotransmitter in the synaptic
cleft may also influence the occupancy of nAChRs causing an additional
activation of the overproduction of glial BP, so that the synaptic system
remains underbalanced. In line with these considerations a biological treatment
of depression should also cope with the overproduction of glial BP, which is
probably caused by mutations in pertinent genes. Therefore, antidepressant
drugs should not only inhibit transmitter reuptake or block postsynaptic
receptors, but they should also be effective on glial nAChRs inhibiting their
overactivation. Otherwise, the synaptic glial-neuronal interaction remains
unbalanced.
In this context it should be mentioned that brain diseases like
Parkinson’s disease also show a lack of transmitter in pertinent synapses, but
this is not necessarily accompanied by a depressive mood. Studies investigating
the frequency of depression in Parkinson’s disease have yielded figures ranging
between 2.7% and 70% (Burn 2002). Hence, depression may only occur if the glial
system is also affected, as I am trying to show.
Although the monoamine deficiency hypothesis of depression is still the
most commonly used model to explain the actions of antidepressant drugs, a
growing body of evidence has accumulated that the hypercholinergic
neurotransmission associated with depressed mood states may be mediated through
excessive neuronal nicotinic receptor activation, and that the therapeutic
actions of many antidepressants may in part be mediated via inhibition of these
receptors (Shytle and others 2002). According to my interpretation of basic
dynamics in tripartite synapses, the cholinergic hypothesis is not
contradictory to the prevailing neurotransmitter hypotheses of depression,
because the synaptic imbalance may essentially depend on the parameters of
glial BP and the presynaptic activation of glial AChRs. In the case of an
excess of ACh in the synaptic cleft, the stimulation of glial nAChRs is
enhanced leading to an additional overexpression of glial BP, so that the
glial-neuronal interaction remains unbalanced despite a cholinergic
hyperactivity. Tricyclic antidepressant drugs are very potent substances, but
are often rejected due to their cholinergic side effects. Do the therapeutic
actions of tricyclic antidepressants concern both the neuronal receptors and
the glial receptors in synapses?
3.3. Mania
Next, which pathophysiological mechanism in tripartite synapses may
cause the manic symptomatology in the sense of an extreme mood elevation (Fig.
8)?
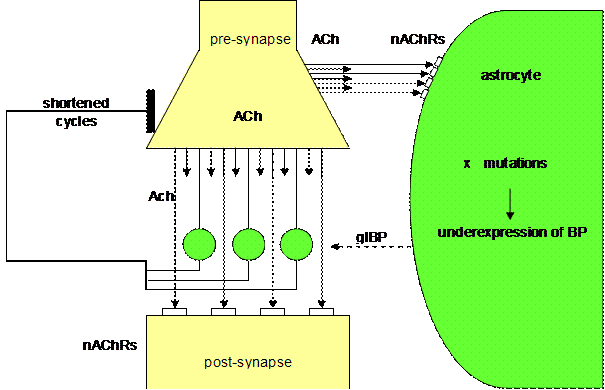
Figure 8. Overbalanced
cholinergic tripartite synapse (Mitterauer, 2004)
As already described, a tripartite
synapse can be overbalanced due to an underexpression of glial BP. This state
may be caused by mutations in genes responsible for the expression of glial BP.
As in depression these mutations arise either spontaneously or by exogenous stress. Unlike the mutations that are hypothesized to
cause depression by increasing glial BP expression, these mutations will result
in reduced BP expression. Under these conditions, there will be a surplus of
neurotransmitter relative to the underexpressed glial BP. In parallel, glial receptors are flooded with
transmitter. This flooding may also
influence the negative feedback mechanism with the effect of shortened cycles
of information processing. Since the glial intentional programs embodied by
glial BP is immediately realizable and “all is appropriate”, a manic patient is
really hypointentional. Dependent on the
transmitter systems or brain areas affected, this synaptic overbalance could
cause the pathophysiology of some manic symptoms like euphoria and feelings of
omnipotence. Additionally, the rapid synaptic cycles could be explanatory of
the manic distractibility, flight of ideas, overactivity and circadian disturbances,
especially insomnia.
The biological treatment of mania focuses on a reduction of the excess
of neurotransmitter in the synaptic cleft. This seemingly leads to a
normalization of information processing, since the occupancy of glial BP
appears to be balanced and glial receptors are not flooded. However, if the
underexpression of glial BP continues after a clinical remission of a manic
episode, hidden symptoms such as loss of motivation, loss of interests and
anhedonia still may persist. This state is often misinterpreted as a depressive
reaction to the manic behavior. Hence, a real remission may only occur if the
genetically determined imbalance of the glial-neuronal interaction in
tripartite synapses is resolved.
3.4. Schizophrenia
Finally, let me try to explain how unbalanced tripartite synapses could
be responsible for the main symptoms of schizophrenia. Schizophrenia, as a syndrome, is composed of
a variety of relatively specific core symptoms. These can be divided into positive
and negative symptoms, with the former including hallucinations, delusions, and
disorganization, and the latter including anergia, flattening of affect, and
poverty of thought content.
If we suppose that mutations in
astrocytes cause the production of non-functional binding proteins in the
synaptic cleft, then the synapses affected are unbalanced. They cannot be
occupied appropriately by their neurotransmitter ligands in the synaptic cleft.
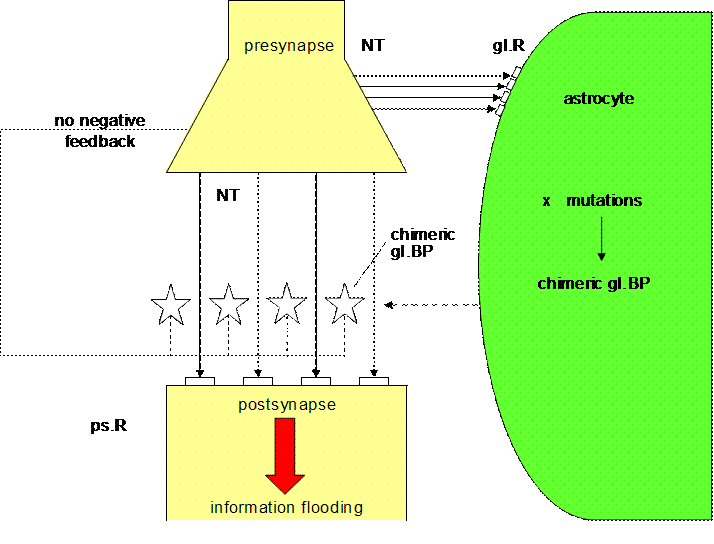
Figure 9. Unbalanced
tripartite synapse (Mitterauer, 2005)
Fig. 9 shows schematically an
unbalanced tripartite synapse. Supposing that genes responsible for the
expression of glial BP are spontaneously or exogenously (stress, etc.) mutated
causing chimeric or non-functional glial BP, then glial BP cannot be occupied
by neurotransmitter (NT). Therefore, the postsynaptic receptors are flooded by
neurotransmitter. In parallel, neurotransmitter occupy glial receptors (gl.R),
activating the production of even more non-functional BP. But that activation
has no effect, since the occupancy of gl.BP does not occur! Most importantly no
negative feedback of the glial system to the presynapse is possible. So the
synaptic information transmission is unconstrained and totally unbalanced. Neuroleptics
can reduce this information overflow by occupying postsynaptic receptors, but
they are unable to influence the molecular mechanisms responsible for the
non-functional gl.BP. If not only gl.BP but also glial receptors are affected,
then the glial-neuronal interaction may break down. Dependent on the brain
areas affected, this state could cause a severe psychobiological disorder like
stuporous catatonia.
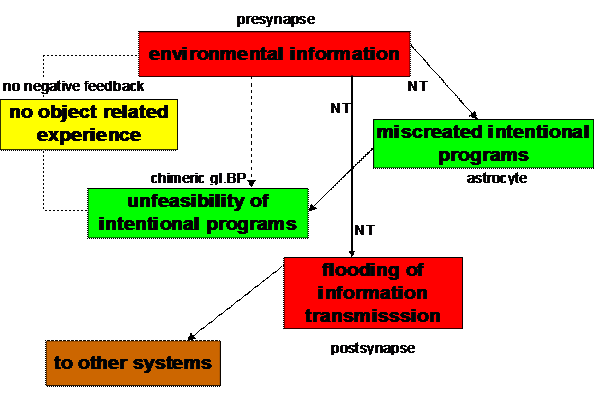
Figure 10. Behavioral
model of miscreated and unfeasible intentional programs in an
unbalanced tripartite synapse
(Mitterauer, 2005)
Interpreted as an elementary behavioral cycle,
glial BPs embody an intentional program that “strives” for realization by means
of neurotransmitter occupancy. An unbalanced tripartite synapse will exist when
non-functional glial BP is unable to bind neurotransmitter and the intentional program is unfeasible. Since
negative feedback is impossible, object related experience does not occur. The
behavior of the system is “obsessed” by a flooding of information transmission
(Fig. 10). Unbalanced tripartite synapses could be characterized as
“dysintentional” because of the unfeasibility of flawed intentional programs.
One could also say that schizophrenic patients are suffering from a weakness of
volition, as already mentioned by Kraepelin (1919). Frith (1999) also refers to
the role of impaired intentions in schizophrenia in the sense of a loss of
awareness of intentions underlying typical symptoms. From a purely biological
point of view, glia have lost their spatio-temporal boundary setting function
in unbalanced tripartite synapses. But what are the consequences of that loss
of glial boundary setting for the interaction between the glial syncytium and
the neuronal networks in the brain?
Let us suppose that glial BP or receptors cannot be occupied and that,
at least locally, neuronal transmission cannot be interrupted. As a result,
neither excitatory nor inhibitory transmitters are able to act in well defined
spatio-temporal functional units within the brain. The type of unbalance of
transmitter systems will depend on the brain areas and the neuronal circuits
affected. A variety of neurotransmitter systems are involved in regulating
information flow via the corticostriato-thalamocortical loops, any of which
could be altered in schizophrenia (Wyatt and others 1995). Concerning dopamine,
a cortico-subcortical imbalance is hypothesized (Davis and others 1991:
Abi-Dargham 2003).
Recent immunohistochemical findings suggest that in the entorhinal and
inferior temporal cortex of the schizophrenic brain the expression of the GABA
(B) receptor is reduced, raising the possibility that GABA (B) receptor
dysfunction is involved in the pathophysiology of schizophrenia (Mizukami and
others 2002; Huntsman and others 1998). Extending these considerations further,
we are not surprised by the fact that alterations in various transmitter
systems have been found in brains of schizophrenic patients, for instance
decreased excitatory synapses in the median temporal lobe (Harrison and
Eastwood 1998; Schmauss 1996). Therefore, it has been hypothesized that the
antipsychotic action of neuroleptic drugs is due to combined neurotransmitter
effects and not to a primary abnormality of dopamine neurotransmission alone
(Johnstone and others 1999; Meltzer 2003).
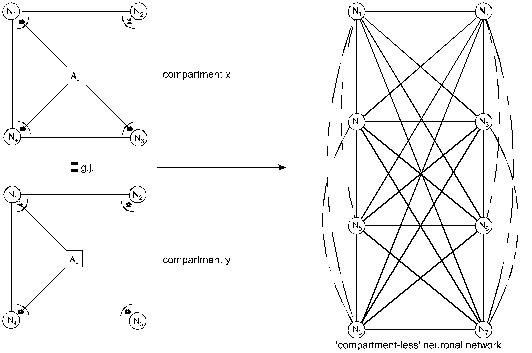
Figure 11. Loss
of glial boundary-setting function and generalization of neuronal
information processing
(Mitterauer, 2003)
A loss of the glial boundary-setting
function in tripartite synapses is depicted in Figure 11. The astrocytes (Aci, Acj) of
compartment x and compartment y are producing non-functional gl.BPs (asterisks)
in the synaptic cleft, so that glia cannot influence neuronal information
processing. This genetically determined disturbance results in a
“compartmentless” neuronal network displayed as a graph of eight neurons with
28 connecting lines (according to the formula (n2-n)/2). Such a
brain is unable to structure the environmental information. One may argue that
a glial determination of neuronal networks into functional units is not
necessary, since the neuronal system is compartmentalized per se (Rall 1995).
However, according to my view, there is a qualitative difference between the
purely neuronal compartments and the glia-determined compartments. Neuronal
compartments are merely functional for information processing, whereas
glial-neuronal compartments may in addition have an information structuring
potency that we need for recognizing the qualitative differences between
objects and individuals in our environment. That capacity may be lost in a
schizophrenic patient. Therefore, one can also speak of a loss of conceptual
boundaries.
Table 1. Interpretation
of basic schizophrenic symptoms
Loss of boundaries Symptoms
Conceptual Thought
disorder
Ontological Delusions
Perceptive Hallucinations
Motoric Catatonic
symptoms
Emotional Affective
flattening
If the loss of boundaries concerns
concepts, a thought disorder results. In the case of a loss of ontological
boundaries among the Self and the others (Non-Selves), delusions occur. This
loss of ontological boundaries can also affect the perception system in the
sense of hallucinations. The loss of boundaries among motor modules shows up in
catatonic phenomena. If the boundaries between emotional qualities are lost,
affective flattening is the typical symptom.
Table 1 shows the basic schizophrenic symptoms (American Psychiatric
Association 1998), which may be caused by a loss of conceptual boundaries. This
disorder can affect cognitive processes like thinking. If a schizophrenic
patient is unable to delimit conceptual boundaries among words, thoughts or
ideas with different meanings, then meaningless word constructs (neologisms) or
disorganized speech are the typical phenomenological manifestations, called “thought
disorder”.
From an ontological point of view, delusions are the consequence of the
loss of boundaries between the Self and the others (Non-Selves). Here, the Self
is defined as a living system capable of self-observation (Mitterauer and Pritz
1978). One could also say that our brain embodies a distinct ontological locus of
self-observation. Everything taking place in the brains of schizophrenic
patients is reality, because they cannot differentiate between their inner
world and the outer world. Therefore, they cannot see ontological differences
between the Self and the Non-Selves. This loss of ontological boundaries may
lead to a delusional misinterpretation of reality.
Hallucinations may be caused by the same disorder. However, the
perception systems are phenomenologically affected. A schizophrenic who hears
the voice of a person in his head is absolutely convinced that this person is
really speaking to him. The loss of ontological boundaries or inner/outer
confusion shows its phenomenological manifestation in the auditory system. Such
a disorder can also occur in other sensory systems.
If the loss of boundaries affects the motor system in the brain, the
symptomatology is called catatonia. A state of catatonic agitation in which a
disinhibited discharge of nearly all motor systems occur, is an expression of
motor generalization with raging and screaming as behavioral components. One
could also say that the brain’s inability to constrain information processing
among motor modules appears in catatonic phenomena. Hence, the catatonic type
of schizophrenia represents a serious disorder of motor behavior. Typical
symptoms are excessive motor activity and motoric immobility (stupor). Both
phenomena appear to be purposeless and not influenced by external stimuli. In
such a catatonic state a patient is unable to communicate. He or she cannot see
the other. Everything that happens, happens in the brain of the patient.
Affective flattening is regarded as a negative schizophrenic symptom
(Dollfus and Petit 1995). This symptom can also be explained as a loss of
boundary setting. The different affective or emotional qualities cannot be produced
within the brain and the communication of feelings is disturbed as a result
(Holden 2003).
My hypothesis of the pathophysiology of schizophrenia might be
consistent, since the pathological brain mechanism described can explain the
main symptoms on the behavioral level. But, in addition we are faced with the
question whether this hypothesis could also be explanatory with concern to
abnormalities of glial cells recently identified in schizophrenic brains. I
think it can.
Let me give an example. Growing evidence for white matter abnormalities
that suggests significant involvement of oligodendroglia in schizophrenia
should be integrated in respect to the glial syncytium and the key role of
astrocytes within. Findings that several genes encoding myelin-related proteins
exhibit consistently reduced expression in schizophrenia may support the
neurodevelopment hypothesis. However, what could be the cause that the onset of
the clinical symptomatology occurs as late as during adolescence or even adulthood?
Supposing that oligodendrocytes do not process axonal information directly, but
that they are dependent on the astrocytic information processing via gap
junctions in the glial syncytium, then the already outlined dysfunctions of
astrocytes may be decisive for the onset of schizophrenic symptomatology.
According to the stress-diathesis model of the etiology of
schizophrenia, environmental stress may activate mutations in astrocytes, since
these cells are actively involved in synaptic information processing, whereas
oligodendrocytes probably are not. As a consequence, one should interpret the
findings of white matter abnormalities in respect to astrocytic dysfunctions.
4. Future prospects
Some final remarks on future
research seem necessary. First of all, the hypotheses proposed here are
experimentally testable. What glial binding proteins concerns, up to now they
have only been identified in lower animals. In addition, a computer based
genome analysis did not find genes that produce these proteins in humans. But I
mistrust such analyses, because I think it is very probable that glial binding
proteins will eventually be identified in vertebrates, including humans. One of
my arguments is this: much of what we know about memory is based on the biochemistry
of Aplysia, the
If the human brain is really not producing glial binding proteins, then
neurotransmitters produced in astrocytes can also achieve a negative feedback
mechanism in tripartite synapses, which already has been experimentally
identified. However, the glial binding protein hypotheses have a greater
explanatory power.
Last but not least, let me shortly comment on the conception of
intentionality or intentional programs, basic for every explanatory model of
human behavior. Recently, I have elaborated a formal, biomimetic model
concerning where and how intentional programs could be generated in our brain.
It can be formally shown that the glial syncytium may be a promising candidate
system, especially with respect to the multifunctionality of gap junctions.
However, this topic would require a separate study.
Considering the structural and functional complexity of the generation
of intentional programs in our brain, I am afraid that experimental brain research
is reaching technical limits. Therefore, I see a real alternative in robotics.
If a biomimetic and formally described brain model is technically implemented
in a robot brain, then the behavior of such a mobile intentional agent teaches
us where we are right and where we are wrong. However, I am convinced that the
agent can only show a human-like behavior if the engineer is able to implement
the glial-neuronal double structure and its glial determined interactions.
References
Abi-Dargham A. 2003. Evidence from
brain imaging studies for dopaminergic
alterations in schizophrenia. In: Kapur S, Lecrubier Y, editors.
Dopamine in
the pathophysiology and treatment of schizophrenia. London: Martin Dunitz.
p 15-47.
American Psychiatric Association, 1998. Diagnostic and statistical manual of
mental disorders. American Psychiatric Association,
Auld DS, Robitaille R. 2003. Glial
Cells and Neurotransmission: An Inclusive
View of Synaptic Function. Neuron 40: 389-400.
Bibring E 1953. The mechanism of
depression. In: Greenacre P, editor.
Affective disorders.
Burn DJ 2002. Beyond the iron mask:
towards better recognition and treatment
of depression associated with Parkinson’s disease. Mov Disord 17:
445-54.
Charles A, Giaume C. 2002.
Intercellular calcium waves in astrocytes:
underlying mechanisms and functional significance. In: Volterra A,
Magistretti PJ, Haydon PG, editors. The tripartite synapse. Glia in synaptic
transmission.
Cooper MS 1995. Intercellular
signalling in neuronal-glial networks.
BioSystems 34: 65-85.
Davis KL, Kahn RS, Ko G, Davidson M. 1991. Dopamine in schizophrenia: a
review and reconceptualization. Am J Psychiatry 148: 1474-86.
Dollfus S, Petit M. 1995. Negative
symptoms in schizophrenia: their evolution
during an acute phase. Schizophrenia
Research, vol. 17. Basic Books, New
Fellin T, Pascual O, Gobbo S et al
2004. Neuronal synchrony mediated by
astrocytic glutamate through activation of extrasynaptic NMDA receptors.
Neuron 43: 729-43.
Frith CD. 1999. The cognitive
neuropsychology of schizophrenia.
Psychology Press.
Gallo V, Ghiani CA 2001. Glutamate
receptors in glia: new cells, new inputs and
new functions. Trends Pharmacol Sci 21: 252-8.
Guenther G. 1963. Das Bewusstsein der Maschinen. Agis-Verlag Baden-Baden.
Harrison PJ, Eastwood SL. 1998.
Prefrontal involvement of excitatory neurons
in medial temporal lobe in schizophrenia. The Lancet 352: 1669-1673.
Hatten ME 1990. Riding the glial
monorail: a common mechanism for glial-guided
migration in different regions of the developing brain. Trends in
Neurosciences
13: 179-184.
Haydon PG. 2001. Glia: Listening and
talking to the synapse. Nature Reviews.
Neuroscience 2: 185-193.
Holden C. 2003. Deconstructing
schizophrenia. Science 299: 333-335.
Huntsman MM,
ratios of alternatively spliced long and short gamma 2 subunit mRNAs of
the
gamma-amino butyrate type A receptor in prefrontal cortex of
schizophrenics. Proc. Natl.
Acad. Sci. USA 95: 15066-15071.
systems. Transactions of the ASME 6.
Johnstone EC, Humphreys MS, Lang FH,
Lawrie SM, Sandler R. 1999
Schizophrenia. University Press,
Kettenmann H, Ransom BR, editors.
1995. Neuroglia.
University Press.
Kimelberg HK, Jalonen TO, Aoki C,
McCarthy K 1998. Transmitter receptor and
uptake systems in astrocytes and their relation to behaviour. In: Laming PR,
Sykova E, Reichenbach A,
Hatton GI, Bauer H, editors. Glial cells: their role
in behaviour.
Kraepelin E. 1971. Dementia praecox
and paraphrenia. Barclay RM, translator.
Kuffler SW, Nicholls JG, Martin AR 1984. Properties and functions of neuroglial cells.
From Neuron to Brain, Sinauer Associates,
Meltzer H. 2003. Multiple
neurotransmitters involved in antipsychotic drug
action. In: Kapur S, Lecrubier Y, editors. Dopamine in the
pathophysiology
and treatment of schizophrenia.
Mitterauer B. 2000a. Clock genes,
feedback loops and their possible roles in the
etiology of bipolar disorders: An integrative model. Medical Hypotheses
55:
155-159.
Mitterauer B. 2000b. Some principles
for conscious robots. Journal of Intelligent
Systems 10: 27-56.
Mitterauer B. 2003. The loss of
self-boundaries: towards a neuromolecular
theory of schizophrenia. BioSystems 72: 209-215.
Mitterauer B. 2004. Imbalance of
Glial-Neuronal Interaction in Synapses: A
Possible Mechanism of the Pathophysiology of Bipolar Disorder. Neuroscientist
10: 199-208.
Mitterauer B 2005. Nonfunctional
glial proteins in tripartite synapses:
a pathophysiological model of schizophrenia. Neuroscientist 11: 192-98.
Mitterauer B, Garvin AM, Dirnhofer R
2000. The sudden infant death
syndrome (SIDS): a neuro-molecular hypothesis. Neuroscientist 6: 154-8.
Mitterauer B, Leitgeb H, Reitboeck H. 1996. The neuro-glial synchronization
hypothesis. Recent Research Development in Biological Cybernetics 1:
137-
155.
Mitterauer B, Pritz WF. 1978. The
concept of the self: a theory of self-
observation. Int. Rev. Psycho-Anal. 5: 179-188.
Mizukami K, Ishikawa M, Hidaka S,
and others. 2002. Immunohistochemical
localization of GABA(B) receptor in the entorhinal cortex and inferior
temporal cortex of schizophrenic brain. Progr. Neuropsychopharmacol.
Biol.
Psych. 26: 393-396.
Newman EA, Zahs KR 1997. Calcium
waves in retinal glial cells. Science 275: 844-6.
Oliet SH, Piet R, Poulain DA 2001.
Control of glutamate clearance and
synaptic efficiency by glial coverage of neurons. Science 292: 923-6.
Post RM 1995. Mood disorders:
somatic treatment. In: Kaplan HJ,
Sadock BJ, editors. Comprehensive textbook of psychiatry.
Williams and Wilkins. P 1152-78.
Rakic P 1988. Specification of
cerebral cortical areas. Science 241: 170-176.
Rall W. 1995. Theoretical
significance of dendritic trees for neuronal input-
output relations. In: Segev I, Rinzel J, Shepherd GM, editors. The
Theoretical
Foundation of Dendritic Function.
Rose CR, Blum R, Pichler B, Lepier
A, and others. 2003. Truncated TrkB-T1
mediates neurotrophin-evoked calcium signalling in glial cells. Nature
426:
74-78.
Schmauss C. 1996. Enhanced cleavage
of an atypical intron of dopamine D3-
receptor pre-mRNA in chronic schizophrenia. J. Neurosci. 16: 7902-7909.
Shytle RD, Silver AA, Lukas RJ, Newman MB,
Sheehan DV, Sanberg PR 2002.
Nicotinic acetylcholine receptors
as targets for antidepressants.
Mol Psychiatry 7: 525-35.
Smit AB, Syed NI, Schaap D, van Minnen J, Klumperman J, Kits KS, and
others. 2001. A glia-derived
acetylcholine-binding protein that modulates
synaptic transmission. Nature
411: 261-268.
Teichberg VI. 1991. Glial glutamate
receptors: likely actors in brain signalling.
FASEB J. 5: 3086-3091.
Virchow R 1846. Über das granulierte Ansehen der Wanderungen
der Gehirnventrikel. Allg. Z. Psychiat. 3: 242-250.
Wyatt RJ, Kirch DG, Egan MF. 1995. Schizophrenia: neurochemical, viral, and
immunological studies.
In: Kaplan, HT, Sadock
BJ, editors. Comprehensive
Textbook of Psychiatry VI, vol. 1. Williams and Wilkins, Baltimore. p
927-
942.
[ BWW Society Home Page ]
© 2006 The BWW Society/The Institute for the Advancement of Positive Global Solutions