Towards
a Comprehensive Pathophysiology of Schizophrenia
Based on Impaired Glial-Neuronal
Interactions
Bernhard J.
Mitterauer
Introduction
Meltzer (2003)
challenged psychiatric research by posing the question how long schizophrenia
will exist as an entity and what will be its future name or names, since this
is diffcult to predict. Here, I propose a pathophysiological model of
schizophrenia that attempts to show that Bleuler’s term may be appropriate.
Disconnections in the neuronal networks in and between areas of brains with
schizophrenia are experimentally well established (Berge and Koenig, 2008). The
underlying disconnection hypothesis states that schizophrenia can be understood
both in cognitive and pathophysiological terms as a failure of proper
functional integration within the brain (Friston, 1998). Accordingly,
schizophrenia may be best understood in terms of abnormal interactions between
different brain regions (Erdi et al., 2007).
However,
structural, molecular and functional changes in glial cells (astrocytes,
oligodendrocytes and microglial cells) have become a major focus of interest in
search for the pathophysiological foundations of schizophrenia. Many studies
show abnormalities in the connecting elements between the nerve cell bodies
(synapses, dendrites and axons) and in all three types of glial cells
(Bernstein et al., 2009). The findings of white matter abnormalities (Di et
al., 2009; Kyriakopoulos et al., 2009) suggest a focus on glial-neuronal
interactions in schizophrenia research. Moreover, a deeper insight into the etiopathophysiology
of this severe disorder is dependent on the underlying brain model from which
the abnormalities are deduced. Schizophrenia is a complex illness that requires
a reappraisal of exclusively neuronal models to include an astrocytic hypothesis
of dysfunction which could lead to a better understanding of schizophrenia on
the behavioral level and could even provide novel treatment strategies
(Steffek, 2007).
Hypotheses
My core
hypothesis is this: astrocytes are equipped with receptors for the various
neurotransmitter substances. The occupancy of these receptors by
neurotransmitters activates the release of gliotransmitters from the astrocyte
modulating neuronal synaptic transmission. This may basically occur by
occupying cognate receptors for gliotransmitters on the presynapse such that
the synaptic information processing is temporarily turned off. If the receptors
on astrocytes are non-functional or cannot even be expressed, an unconstrained
synaptic flux arises. Since each astrocyte via its processes contacts a
distinct amount of synapses generating a glial-neuronal domain, in the case of
non-functional astrocytic receptors the domain organization decomposes. Hence,
the basic pathophysiological fault of schizophrenia may be caused by synaptic
gaps between the neuronal components and the astrocyte.
In the acute
delusional stage of the illness no significant network impairments may occur,
but the functional separation between the neuronal and glial networks on the
synaptic level may lead to uncontrolled synaptic information processing. Since
the astrocyte domain organization is interrupted, the information processing
generalizes in both networks which may be responsible for delusional
misinterpretations of a given reality in the environment. In parallel, the
unconstrained synaptic transmitter flux may impair oligodendrocyte-axonic
interactions leading to the decay of both oligodendroglia and axons which may
explain the cognitive and emotional defects of schizophrenics in the
progressive stage of the illness. Supposing that in the glial networks
intentional programs may be generated which are tested in the neuronal networks
regarding their feasibility in the environment, a brain with schizophrenia is unable
to do so because of the synaptic separation between the neuronal and glial
networks. One can speak of schizophrenic dysintentionality.
What
non-schizophrenic delusions concern, these may be based on a different
pathophysiology. Astrocytes also exert a modulating function in extrasynaptic
neurotransmission via the activation of inhibitory interneurons that negatively
feed back to the presynapse. If, for various reasons (e.g. stress), a synaptic
neurotransmitter flooding occurs, although the astrocytic receptors are
normally expressed an unconstrained information flux as in schizophrenic
delusions may occur, but these are caused by a disturbance of the extrasynaptic
negative feedback mechanism. Before further elaborating on these hypotheses,
the underlying brain model will be outlined.
Brain model of glial-neuronal interactions
Description of the networks
The proposed
biological brain model is based on glial-neuronal interactions (Mitterauer,
1998; 2007). The nervous tissue of the brain consists of the neuronal system
(neurons, axons, dendrites) and the glial system (astrocytes, oligodendrocytes
with myelin sheaths enfolding axons, radial glia, and microglia). Experimental
results are inspiring a major reexamination of the role of glia in the
regulation of neural integration in the central nervous system (Kettenmann and
Ransom, 2005). Figure 1 shows a schematic diagram of the glial-neuronal
interaction: two astrocytes (Ac1,2) are shown in this very simple
model, whereby in each case only one neuron (N1,2) belonging to an
astrocyte is taken into consideration. Halassa et al. (2007) identified how a
single astrocyte contacts only four to eight neurons, but 300 to 600 synapses
via its processes. The glial network (syncytium) consists in this schema of two
astrocytes and two oligodendrocytes (Oc1,2) interconnected via gap
junctions (g.j.). The neuronal system shows two neurons (N1,2) with
two afferent axons (Axi,j) and two afferent axodendritic synapses
(Sai,j), two efferent axons (Ax1,2) with myelin sheaths
(Ms) and a node of
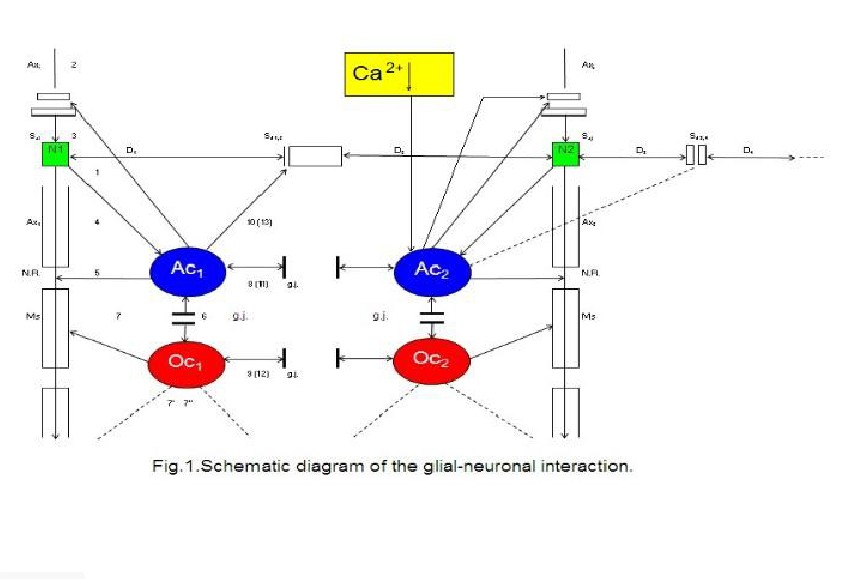
Two astrocytes (Ac1, 2) are
shown in this very simple model, whereby in each case only one neuron belonging
to an astrocyte is taken into consideration. The glial network (syncytium)
consists of two astrocytes and two oligodendrocytes (Oc1, 2)
belonging to them. Gap junctions (g.j.) exist between the astrocytes and the
oligodendrocytes. The neuronal system shows two neurons (N1, 2) with
two afferent axons (Axi, j) and two afferent axo-dendritic synapses
(Sai, j), two efferent axons (Ax1, 2) with myelin sheaths
(Ms) and a node of Ranvier (N.R.), as well as two dendro-dendritic synapses (Sd1
, 2¸3, 4) with the corresponding dendrites (D1, D2,
D3, D4). A decrease of calcium (Ca2+) in the
extrasynaptic space (ex sy space) activates astrocytes that negatively feed
back to the synapse (Rusakov, 2012).
Ranvier (N.R.),
as well as two dendro-dendritic synapses (Sd1,2,3,4) with the
corresponding dendrites (D1,D2;D3D4).
In addition, a decrease of calcium (Ca2+) in the extrasynaptic space
(exsyspace) activates astrocytes that negatively feed back to the synapse
(Rusakov, 2012).
Outline of an
astrocytic syncytium
Different
connexins allow communication between diverse cell populations or segregation
of cells into isolated compartments according to their pattern of connexin
expression. Gap junctions are composed of hemichannels (connexons) that dock to
each other via their extracytoplasmic extremities. Each hemichannel is an
oligomer of six connexin proteins (Cx). In the central nervous system,
cell-specific and developmentally regulated expression of eight connexins has
been demonstrated (Dermietzel and Spray, 1998).
My
hypothesis focuses on gap junctions between astrocytes, the main glial cell
type besides oligodendrocytes and microglia. Gap junctions are considered to
provide a structural link by which single cells are coupled to build a
functional syncytium with a communication behavior that cannot be exerted by
individual cells. Gap junctions of an astrocytic syncytium consist of the four
identified connexins Cx43, Cx32, Cx26 and Cx45, forming homotypic (i.e. gap
junction channels formed by hemichannels of the same kind) and heterotypic gap
junction channels (i.e. formed by hemichannels of different kinds). Whereas
astrocytes are interconnected with their neighbors via gap junctions, the
interactions of astrocytes with neurons occur mainly in synapses called
tripartite synapses (Araque et al., 1999).
Figure
2 shows a diagrammatic scheme depicting an astrocytic syncytium. Six astrocytes
(Ac1…Ac6) are completely interconnected via fifteen gap
junctions (g.j.) according to the formula n:2 (n-1). Each astrocyte contacts a
neuronal synapse, building a tripartite synapse in the sense of a
glial-neuronal unit. Admittedly, this simple diagram refers only to the
elementary components and their connections in an astrocytic syncytium.
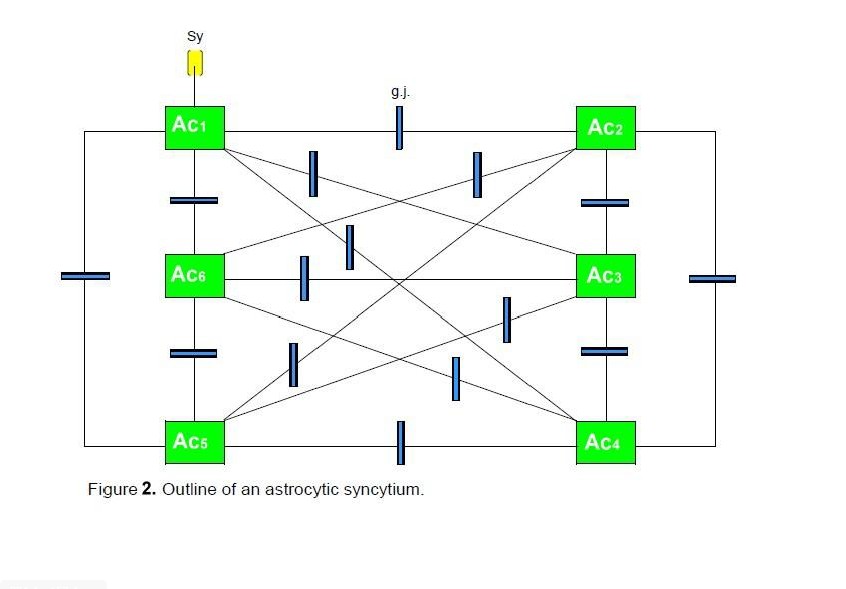
Six
astrocytes (Ac1...Ac6) are interconnected via 16 gap
junctions (g.j.) building a complete syncytium. Each astrocyte contacts a
neuronal synapse representing a tripartite synapse (for the sake of clarity,
only one synaptic contact [Sy] is shown).
Since
in the brain each macroscopic gap junction is an aggregate of many, often
hundreds of tightly packed gap junction channels are observed (Ransom and Ye,
2005). The number and composition of gap junctions can be dynamically regulated
at the level of the endoplasmic reticulum by either upregulating connexin
biosynthesis or decreasing the rate of connexin degradation, and at the cell
surface by enhancing gap junction assembly or reducing connexin degradation.
Model of a
glutamatergic tripartite synapse
The
close morphological relations between astrocytes and synapses as well as the
functional expression of relevant receptors in the astroglial cells prompted
the appearance of a new concept known as the tripartite synapse, and which I
call glial-neuronal synaptic unit (GNU). Araque et al (1999) showed that glia
respond to neuronal activity with an elevation of their internal Ca2+ concentration
which triggers the release of chemical transmitters from glia themselves, and,
in turn, causes feedback regulation of neuronal activity and synaptic strength.
Although a true understanding of how the astrocyte interacts with neurons is
still missing, several models have been published (Halassa et al., 2009). Here,
I focus on a modified model proposed by Newman (2005). Figure 3 represents the interaction of the
main components of synaptic information processing as follows: sensori-motoric
networks compute environmental information activating the presynapse (1). The
activated presynapse releases glutamate (GL) from vesicles (v) that occupy both
postsynaptic receptors (poR) and receptors on the astrocyte (acR) (2) (For the
sake of clarity, only one receptor is shown). Moreover, glutamate may also
activate gap junctions (g.j.) in the astrocytic syncytium leading to an
enhanced spreading of Ca2+ waves (3). In parallel, the occupancy of
the astrocytic receptors by glutamate also activates Ca2+ within the
astrocyte (4). This mechanism exerts the production of glutamate (5) and
adenosine triphosphate (ATP) (6) within the astrocyte, now functioning as
gliotransmitters. Whereas the occupancy of extrasynaptic pre- and postsynaptic
receptors by glutamate is excitatory (7), the occupancy of these receptors by
ATP is inhibitory (8). In addition, neurotransmission is also inactivated by
the reuptake of glutamate in the membrane of the presynapse mediated by
transporter molecules (t) (9). Most importantly, ATP inhibits the presynaptic
terminal via occupancy of cognate receptors, temporarily turning off synaptic
neurotransmission in the sense of negative feedback (10). Finally, synaptic
information processing is transmitted to neuronal networks that can activate
the synapse again (11).
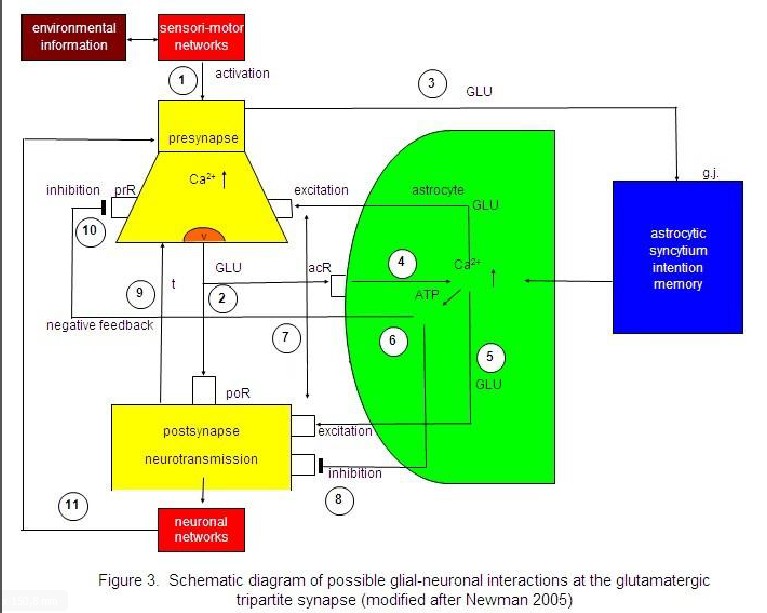
Sensori-motor
networks compute environmental information activating the presynapse (1). The
activated presynapse releases glutamate (GLU) from vesicles (v) occupying both
postsynaptic receptors (poR) and receptors on the astrocyte (acR) (2). GLU also
activates gap junctions (g.j.) in the astrocytic syncytium, enhancing the
spreading of Ca2+ waves (3). In parallel, the occupancy of acR by
GLU also activates Ca2+ within the astrocyte (4). This mechanism
exerts the production of GLU (5) and adenosinetriphosphate (ATP) (6) within the
astrocyte, now functioning as gliotransmitters. Whereas the occupancy of the
extrasynaptic pre- and postsynaptic receptors by GLU is excitatory (7), the
occupancy of these receptors by ATP is inhibitory (8). In addition,
neurotransmission is also inactivated by the reuptake of GLU in the membrane of
the presynapse mediated by transporter molecules (t) (9). ATP inhibits the
presynaptic terminal via occupancy of cognate receptors (prR) temporarily
turning off synaptic neurotransmission in the sense of a negative feedback
(10). Synaptic information processing is transmitted to neuronal networks
activating the synapse again (11).
In
spite of evidence that astrocytes release glutamate by a Ca2+-dependent
vesicule mechanism that resembles release from neurons, important differences
between glial and neuronal release exist. Glutamate release from astrocytes
occurs at a much slower rate than does release from neurons, and it is probably
triggered by smaller increases of cytoplasmic Ca 2+. Importantly, we
apparently deal with different time scales of presynaptic and astrocytic
glutamate release. Here, the astrocytic modulatory function of synaptic neurotransmission
may occur within seconds or minutes (Stellwagen and Malenka, 2006). I hypothesize that the duration from
presynaptic activation to the inhibition of synaptic neurotransmission may also
be dependent on the amount of astrocytic receptors that must be occupied by
glutamate. This mechanism may be based on the occupancy probability of
astrocytic receptors by glutamate releases from the presynaptic terminal. In
addition, the release of ATP from astrocytes may also be dependent on a
comparable mechanism. Accordingly, in GNU’s glia may have a temporal
boundary-setting function in temporarily turning off synaptic transmission
(Auld and Robitaille, 2003; Mitterauer, 1998).
Outline of an astrocyte domain organization
In all mammals,
protoplasmic astrocytes are organized into spatially non-overlapping domains
that encompass both neurons and vasculature. An astrocyte domain defines a
contiguous cohort of synapses that interacts exclusively with a single
astrocyte. Synapses within a particular territory are thereby linked via a
shared astrocyte partner, independent of a neuronal networking (Oberheim et
al., 2006). Single protoplasmic astrocytes operate as a “Lokal Hub” (Pereira
and Furlan, 2010). Figure 4 shows an outline of an astrocyte domain organization.
An astrocyte (Acx) contacts the synapses (Sy) of four neurons (N1…N4)
via its processes (P1…P4). Each process is equipped with
one to four receptor qualities (Rq). For example, P1 contacts the
synapses of N2 exclusively via its receptors of quality a. P2
has already two receptor qualities available (a, b), P3 three
receptor qualities (a, b, c) and P4 is able to contact the synapses
of N1 via four receptor qualities (a, b, c, d). Astrocyte (Acx)
is interconnected with another astrocyte (Acy) via gap junctions
(g.j.) forming an astrocytic network (syncytium). The neurons per se are also
interconnected (neuronal network).
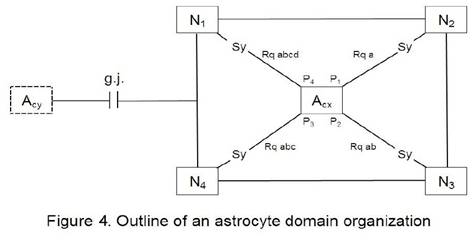
An astrocyte (Acx)
is interconnected via four processes (P1…P4) with the
synapses (Sy) of four neurons (N1…N4). Each process is on
its endfoot equipped with receptors for the occupancy with neurotransmitters
according to a combinational rule (Mitterauer, 2010). As an example, the
receptor P1 contacting N2 embodies only one receptor
quality (Rqa). P2 contacts N3 with two different receptor
qualities (Rqab). P3 contacts N4 with Rqabc and P4
contacts N1 with Rqabcd. This simple diagram represents an astrocyte
domain. Astrocyte (Acx) is interconnected with Acy via
gap junctions (g.j.) as shown in more detail in Figure 2.
It is
experimentally verified that astrocytes can express almost all receptors for
important transmitter systems. In certain cases, individual astroglial cells
express as many as five different receptor systems linked to Ca2+
mobilization (Kettenmann and Steinhäuser, 2005). Each astrocyte territory
represents an island made up of many thousands of synapses (about 140.000 in
the hippocampal region of the brain, for instance), whose activity is controlled by that
astrocyte (Santello and Volterra, 2010). On the average, human astrocytes
extend 40 large processes radially and symmetrically in all directions from the
soma so that each astrocyte supports and modulates the function of roughly two
million synapses in the cerebral cortex (Oberheim et al., 2006). Astrocytic
receptors are mainly located on the endfeet of the processes. Here, we
apparently deal with a high combinational complexity of astrocyte-synaptic
interactions.
Oligodendrocyte-Axonic Interactions
For normal brain
function it is essential that signals pass rapidly between neurons.
Oligodendrocytes play an important role in assuring fast neuronal signaling in
the central nervous system. By covering neuronal axons with myelin which
decreases the effective axonal membrane capacity, they reduce the charge needed
to depolarize the axon and hence allow the action potential to travel much
faster by saltatory conduction from one node of Ranvier to the next.
During
development, oligodendrocytes are generated from precursor cells. These
initially differentiate into immature cells that put out processes seeking
axons to myelinate, and eventually they form mature cells with parallel
processes myelinating up to 30 different axons. The production of myelinated
axons requires a precise matching of the number of oligodendrocytes generated
to the length of axons to be myelinated. This may be regulated in part by
neurotransmitter receptors activated by substances released by active axons.
Such interactions may also be important for maintaing the myelination of mature
axons. Thus, neurotransmitter receptors play an important role in the life and
death of oligodendrocytes (Káradóttir and Attwell, 2007).
Impulse activity
in axons affects the development of oligodendrocytes, and thus myelination.
Some of the effects of impulse activity inhibit myelination and some stimulate
it. Here, the question arises as to how oligodendrocytes know which axons are
electrically active. Three mechanims have been identified that regulate
myelination or the development of myelin forming glia in response to electrical
stimulation of axons in vitro (Fields, 2010). Specific frequencies of
electrical impulses control the amount of L1CAM present on unmyelinated axons,
a cell adhesion molecule that is necessary for myelination (Stevens et al,
1998). The neurotransmitter adenosine 5’-triphosphate (ATP) is released from
axons and activates receptors on astrocytes, causing them to release the
leukemia inhibitory factor that stimulates myelination by mature
oligodendrocytes (Ishibashi et al, 2006). Adenosine derived from hydrolysis of
released ATP promotes oligodendrocyte precursor cell development and thus
increases myelination (Stevens et al, 2002). Moreover, a nonsynaptic mechanism
for ATP release from axons has also been identified (Fields and Ni, 2010).
Glial modulating
function in extrasynaptic neurotransmission
Rapid signal
exchange between astroglia and neurons across the interstitial space emerged as
an essential element of synaptic circuit functioning in the brain (Rusakov,
2012; De-Miguel and Fuxe, 2012). The excitatory neurotransmitter glutamate and
the inhibitory neurotransmitter GABA are thought to be the basic transmitters
that are regularly from the presynapse occupying cognate receptors on the
surface of astrocytes (Auld and Robitaille, 2003). Recently, extracellular
calcium ions (Ca2+) have been identified that can report neuronal
activity to astroglia (Torres et al., 2012). Astrocytes in the hippocampus can
respond to activity-induced partial Ca2+ depletion in the
extracellular space by generating prominent intracellular waves. The underlying
Ca2+ sensing mechanism may involve the opening of the hemichannel
connexin 43 in the astrocytic syncytium which in turn triggers the release of
adenosine triphosphate (ATP), enhancing the activity of inhibitory
interneurons, thus potentially exerting negative feedback. This mechanism
significantly reduces excessive excitatory activity of neuronal circuits
(Figure 5).
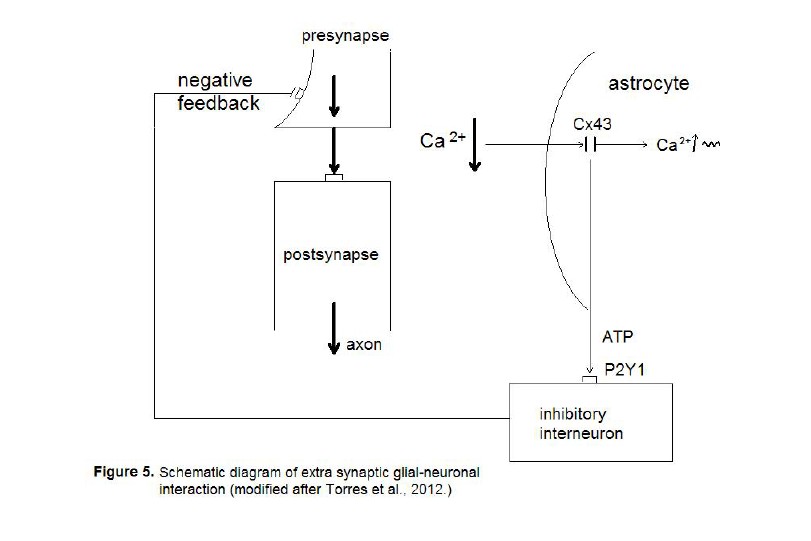
The neurotransmission from the presynapse
to the postsynapse is hyperexcited (fat arrows) leading to partial depletion of
calcium in the extrasynaptic space (Ca 2+ ↓). The astrocyte
senses the imbalance via connexin 43 (Cx43) hemichannels leading to an increase
of intracellular calcium waves (Ca 2+ ↑ ~). In parallel, the
astrocyte produces adenosine-triphosphate (ATP) that activates P2Y1 receptors
on inhibitory interneurons that negatively feed back to the presynapse.
Here we deal with
a new mechanism of the glial temporal boundary setting function (Mitterauer,
1998). Whereas astrocytes are capable of turning off synaptic neurotransmission
by occupying receptors with gliotransmitters, in extrasynaptic neurotransmission
an indirect pathway activating inhibitory interneurons through astrocytic ATP
release is at work exerting a negative feedback on the presynapse. This
mechanism may play an important role in disorders with synaptic
hyperexcitability as epilepsy or even non-schizophrenic delusions (see below).
Pathophysiological model of schizophrenia
The
core symptoms of schizophrenia can be divided into positive and negative
symptoms, with the former including hallucinations, delusions, and
disorganization, and the latter including anergia, flattening of affect, and
poverty of thought content accompanied by significant disturbances in cognitive
function (Meltzer, 2003). Hypotheses concerning the etiology of schizophrenia
comprise biological, psychological and sociological approaches (Carpenter and
Buchanan, 1995; Shastry, 2002; Kapur and Lecrubier, 2003; Lenzenweger et al.,
2007). Generally one can explain delusions and hallucinations in terms of a
“loss of ego- or self-boundaries in the sense of an inner/outer confusion”
(Fisher and Cleveland, 1968; Sims, 1991; Mitterauer, 2003).
Non-functional
astrocytic receptors cause astrocytic domain decomposition
Let me
attempt to show how it may be possible to deduce the main schizophrenic
symptoms from an unbalanced tripartite synapse. If the glial receptors are
totally non-functional and therefore cannot be occupied by neurotransmitters,
the system is unbalanced. As in Fig. 6 depicted, the glial receptors (glR) are
non-functional (crosses) and cannot be occupied by neurotransmitters (NT), so
that the activation of the gliotransmitters (GT) is impossible. Hence, they
cannot negatively feedback to the receptors on the presynapse (prR) and are
unable to depolarize the postsynaptic neuron. As a consequence, the glia lose
their inhibitory or boundary-setting function and the neural transmitter flux
is unconstrained, as the flux of thought on the phenomenological level.
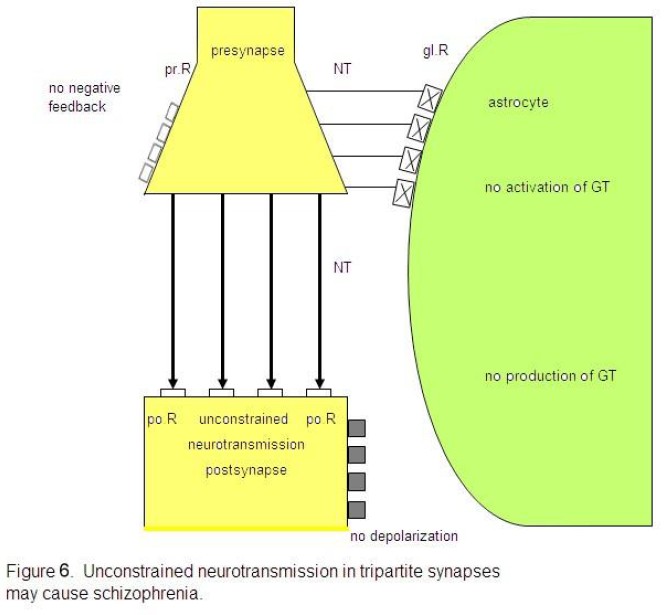
Non-functional glial receptors (glR),
depicted by crosses, cannot be occupied by neurotransmitters (NT). Since the
activation and production of gliotransmitters (GT) is not possible, glia do not
negatively feed back to the presynaptic receptors (prR) and cannot depolarize
the postsynaptic neuron. This severe synaptic disturbance leads to an
unconstrained neurotransmission (fat arrows).
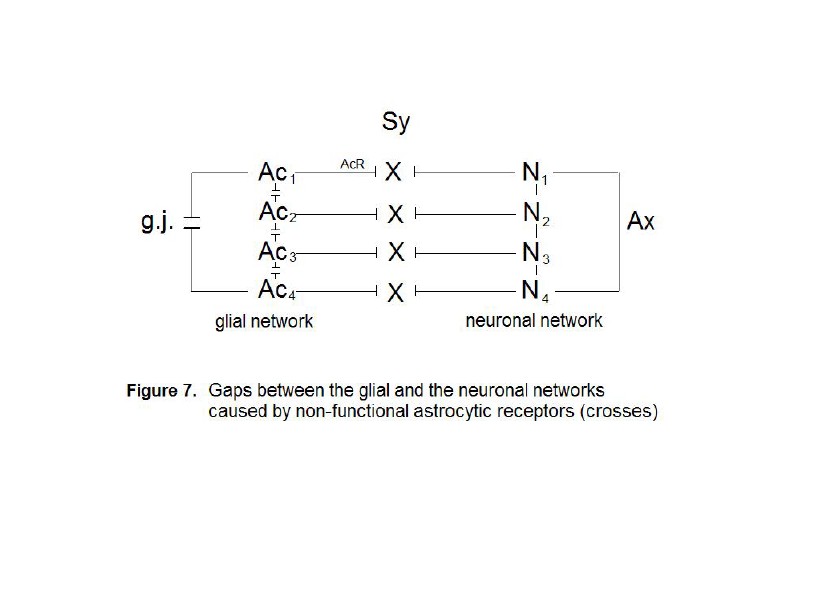
Four astrocytes (Ac1...Ac4)
are interconnected via gap junctions (g.j.) building a glial network. Four
neurons (N1...N4) are interconnected via axons (Ax)
building a neuronal network (dendrites not shown). The synaptic interactions
between the glial and neuronal networks are interrupted, since the astrocytic
receptors (AcR) are nonfunctional (crosses).
As already
described, synaptic glial-neuronal interactions are organized into astrocyte
domains. Non-functional astrocytic receptors decompose the glial network from
the neuronal one causing a gap between the two. Thus, the term schizophrenia is
correct in view of this pathophysiological model. Fig. 7 represents a simple
diagram of this network decomposition. Four astrocytes (Ac1…Ac4)
are interconnected by gap junctions (g.j.) building a glial network
(syncytium). Four neurons (N1…N4) are also interconnected
via axons (Ax) generating a neuronal network. Since the astrocytic
receptors (AcR) are non-functional, synaptic glial-neuronal
interactions are disrupted (crosses).
Because the
astrocyte domain organization may be significant for qualitative information
structuring in the brain (Mitterauer, 2010), its decomposition leads to a
generalization of information processing in the neuronal networks. Thus,
schizophrenics cannot recognize qualitatively different features of subjects
and objects but instead they must think in general cases. This cognitive
incapability may cause a misinterpretation of a given reality in the sense of
delusions and hallucinations. One may argue that a glial
determination of neuronal networks into functional units is not necessary
because the neuronal system is compartmentalized per se
(Rall, 1995). However, according to my view, there is a qualitative difference
between the purely neuronal compartments and the glia-determined domains.
Neuronal compartments may be merely functional for information processing,
whereas glial-neuronal compartments may in addition have an
information-structuring potency that we need for recognizing the qualitative
differences between objects and individuals in our environment. That capacity
may be lost in schizophrenic patients. Therefore, one can also speak of a loss
of conceptual boundaries in schizophrenia. Let me now deduce the main symptoms
of schizophrenia from unbalanced tripartite synapses caused by non-functional
glial receptors. Table 1 shows the basic schizophrenic symptoms (American
Psychiatric Association, 1998) that may be caused by a loss of conceptual
boundaries. This disorder can affect cognitive processes such as thinking. If a
schizophrenic patient is unable to delimit conceptual boundaries among words,
thoughts, or ideas with different meanings, then meaningless word constructs
(neologisms) or disorganized speech are the typical phenomenological
manifestations, called “thought disorder”.
Table 1. Interpretation of basic schizophrenic symptoms
|
Loss of boundaries |
Symptoms |
|
Conceptual |
Thought disorder |
|
Ontological |
Delusions |
|
Perceptive |
Hallucinations |
|
Motoric |
Catatonic symptoms |
|
Emotional |
Affective flattening |
If
the loss of boundaries concerns concepts, a thought disorder results. In the
case of a loss of ontological boundaries among the Self and the others
(Non-Selves), delusions occur. This loss of ontological boundaries can also
affect the perception system in the sense of hallucinations. The loss of
bundaries among motor modules shows up in catatonic phenomena. If the
boundaries between emotional qualities are lost, affective flattening is the
typical symptom. (Mitterauer, 2003)
From
an ontological point of view, delusions are the consequence of the loss of
boundaries between the self and the others (nonselves). Here, the self is
defined as a living system capable of self-observation (Mitterauer and Pritz,
1978). One could also say that our brain embodies a distinct ontological locus
of self-observation. Everything taking place in the brains of schizophrenic
patients is reality because they cannot differentiate between their inner world
and the outer world. Therefore, they cannot see ontological differences between
the selves and the nonselves. This loss of ontological boundaries may lead to a
delusional misinterpretation of reality.
Hallucinations
may be caused by the same disorder. However, the perception systems are
phenomenologically affected. A schizophrenic who hears the voice of a person in
his head is absolutely convinced that this person is really speaking to him.
The loss of ontological boundaries or inner/outer confusion shows its
phenomenological manifestation in the auditory system. Such a disorder can also
occur in other sensory systems.
If the
loss of boundaries affects the motor system in the brain, the symptomatology is
called catatonia. A state of catatonic agitation in which a disinhibited
discharge of nearly all motor systems occurs is an expression of motor
generalization with raging and screaming as behavioral components. One could
also say that the brain’s inability to constrain information processing among
motor modules appears in catatonic phenomena. Hence, the catatonic type of
schizophrenia represents a serious disorder of motor behavior. Typical symptoms
are excessive motor activity and motoric immobility (stupor). Both phenomena
appear to be purposeless and not influenced by external stimuli. In such a
catatonic state, a patient is unable to communicate. He or she cannot see the
other. Everything that happens takes place in the brain of the patient.
Affective
flattening is regarded as a negative schizophrenic symptom (Arajärvi et al.,
2006). This symptom can also be explained as a loss of boundary setting. The
different affective or emotional qualities cannot be produced within the brain,
and the communication of feelings is disturbed as a result (Holden, 2003).
Some
genetic considerations
There
is growing evidence of disease-related altered astrocyte gene expression. These
findings suggest an imbalance of glutamate-glutamine cycle in the communication
of neurons and astrocytes (Hashimoto et al., 2005). There is also evidence for
a broad involvement of astrocytes in other aspects of the pathophysiology in
schizophrenia (Bernstein et al., 2009). However, if we focus on nonfunctional
receptor proteins on astrocytes, an aberrant splicing may represent a genetic
candidate mechanism. Aberrant or non-splicing causes truncated or chimeric
proteins such that receptor occupancy is not possible. What the mechanisms of
aberrant splicing concerns, significant findings have been reported (Faa et
al., 2010). Nonsense, missense, and even synonymous mutations can induce
aberrant skipping of the mutant exon, producing nonfunctional proteins. If the
exchange of nucleotides generates a synonymous codon that represents the same
aminoacid as the original triplet, one speaks of a silent mutation. These
mutations have erroneously been classified as nonpathogenic, but are now
recognized as affecting the splicing machinery resulting in defective proteins.
Aberrant splicing may play a decisive role in the pathophysiology of various
diseases (Wang and Cooper, 2007). Why not in schizophrenia, as I hypothesized
nearly a decade ago?
Recently,
gene losses in the human genome have been identified (Quintana-Murci, 2012).
These loss-of-function variants are located in human protein-coding genes.
Since the first comprehensive, genome-wide catalogue of variants likely to
disrupt protein-coding genes is now available, this genetic approach could also
be promising for the identification of nonfunctional receptors on astrocytes in
brains with schizophrenia. Moreover, epigenetic factors may also play an
important role. Epigenetics broadly refers to heritable changes in phenotype or
gene expression caused by mechanisms other than changes in the underlying
primary DNA sequence. Several major types of epigenetic mechanisms are DNA
methylation, genomic imprinting, histone modifications, and expression control
by noncoding RNA. Recent data suggest the influence of these epigenetic
alterations in schizophrenia (Deng et al., 2010).
Unconstrained
neurotransmission may cause demyelination
Generally,
in pathological conditions neurotransmitters can be released excessively,
damaging the cells they normally act on. Since oligodendrocytes have receptors
for the various transmitters, neurotransmitter excess can cause demyelination
(Káradóttir and Attwell, 2007). Already in 1977 a study was published
documenting severe breakdown of myelin in dogs injected with myelin (Saakov et
al, 1977). In the grey matter of the brain the death of neurons in pathological
conditions is often caused by a rise of extracellular glutamate concentration
activating NMDA receptors and causing an excessive rise of Ca2+.
Glutamate can also damage white matter oligodendrocytes. Concentrations of
glutamate which alone are not toxic sensitize oligodendrocytes to subsequent
complementary attack that inserts membrane attack complexes into the
oligodendrocyte, allowing a toxic Ca2+ influx to occur (Alberdi et
al, 2006).
In
line with the presented synaptic model of the pathophysiology of schizophrenia,
an unconstrained flux of neurotransmitters occurs. This may hold for all the
various neurotransmitter types. This unconstrained flux of neurotransmitters
may affect oligodendrocytes either by flooding of their cognate receptors on
oligodendrocytes, exerting a toxic Ca2+ influx, or via a
hyperactivation of axons with an excess of axonic ATP production and a
consequent toxic effect on oligodendrocytes (Mitterauer and Kofler-Westergren,
2011) (Fig. 8). In addition, ATP released from axons cannot activate astrocytic
receptors, since they do not function. Therefore, a stimulation of myelination
by mature oligodendrocytes is not possible. These pathological mechanisms may
cause demyelination, as observed in brains with schizophrenia (Skelly et al,
2008; Takahashi et al, 2010). Note, although decreased expression of
oligodendrocyte-related genes has been identified (Höstad et al, 2009), it
seems implausible that genetics alone could account for demyelination in
schizophrenia (Fields, 2009).
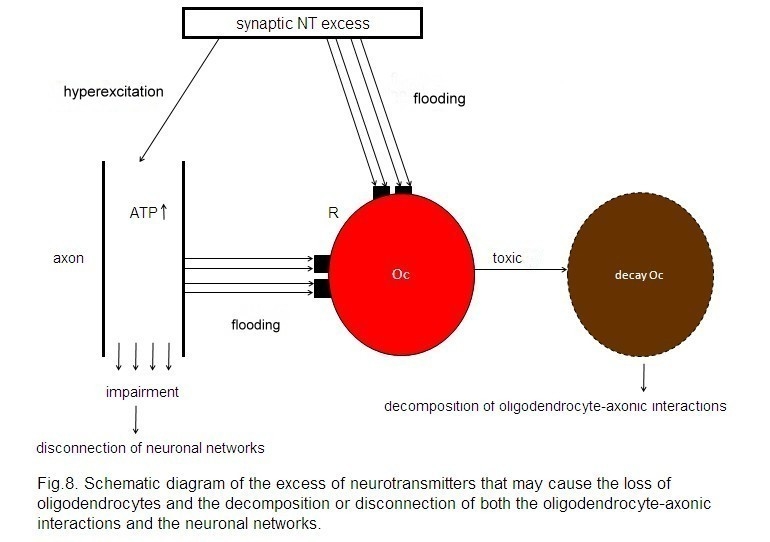
A synaptic neurotransmitter (NT) excess
hyperexcites the axon and floods the cognate receptors (R) on the
oligodendrocyte (Oc). In parallel, a non-synaptic ATP excess occurs, also
flooding R. This flooding of NT and ATP exerts a toxic effect on the Oc,
leading to its decay. These mechanisms may be responsible for the decomposition
of oligodendrocyte-axonic interactions and the disconnection of neuronal
networks.
Moreover,
the permanent hyperactivation of axons may also impair them so that neuronal
networks disconnect. This may represent a possible mechanism that could at
least in part be responsible for the disconnection of neuronal networks
identified in brains with schizophrenia (Höstad et al, 2009).
Loss of
Oligodendrocytes May Cause a Decay of Gap Junctions in the Panglial Syncytium
Responsible for Memory Impairment
Since
gap junctions between astrocytes and oligodendrocytes are heterotypic, composed
of special astrocytic connexins and different oligodendrocytic connexins, a
loss of oligodendrocytes disrupts astrocyte-oligodendrocytic gap junctions in
the panglial syncytium, leading to a “leaky” syncytium (Fig. 9). Gap junctions
form plaques that may embody memory structures (Robertson, 2002). Therefore, a
loss of gap junctions may impair memory. In addition, genetic or (and)
exogenous disorders can affect gap junctional plaque formation which may be the
case in schizophrenia. In this context one could speak of a syncytiopathy in
schizophrenia (Mitterauer, 2009). However, memory impairment may also be caused
by a disorganization of neuronal networks (Wolf et al, 2008).
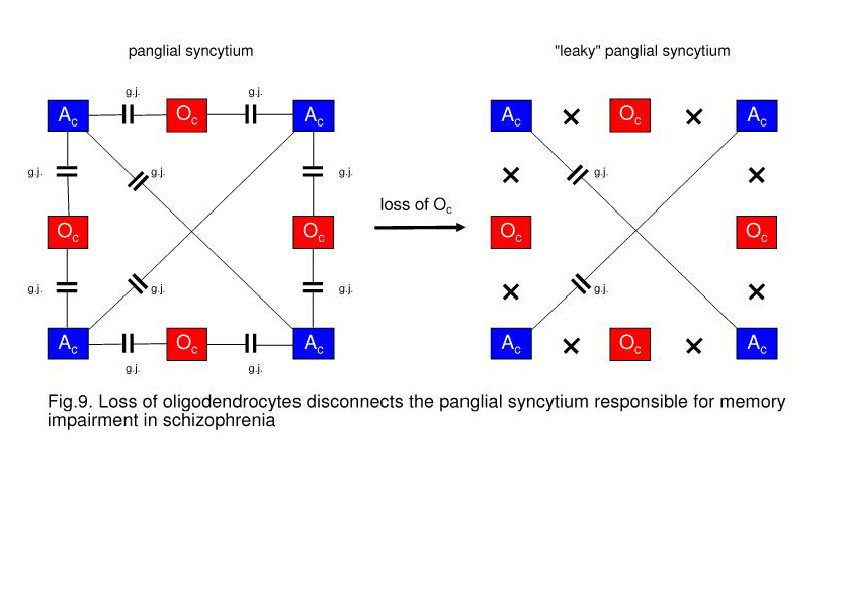
On the left, a panglial syncytium composed
of astrocytes (Ac) and oligodendrocytes (Oc) interconnected via gap junctions
(g.j.). A loss of Oc may cause a decay of Oc-Ac gap junctions (crosses). Such a
“leaky” panglial syncytium may be responsible for memory impairment.
Schizophrenic
dysintentionality based on a severe disorder of glial-neuronal interactions
According
to Frith (1992), schizophrenia can be explained by a failure to integrate the
intention to act with the perceptual registration of the consequences of that
action. At a neurobiological level this integrative abnormality might
correspond to a failure to integrate signals from the (intentional) prefrontal
regions and the (perceptual) temporal cortices. However, this is a pure
neuronal view excluding the glial cell system. In further elaborating my theory
of glial-neuronal interaction, I have hypothesized that the intentional or
action programs of the brain may be generated in the panglial syncytium
(Mitterauer, 2007). Based on a formal model it can be shown how glial syncytia
compute in a highly combinatorial manner cycles of various lengths via gap
junctions. These cycles are transferred in tripartite synapses to the neuronal
system (Mitterauer, 2011). The neuronal system tests these intentional programs
with regard to their feasibility in the environment. In feeding back the
feasibility of intentional programs to the glial syncytium, learning processes
can occur.
For
the clinically experienced psychiatrist it is evident that patients with
schizophrenia are unable both to test their delusional programs and to realize
these unrealistic intentions in the environment. I have named this disorder
“schizophrenic dysintentionality” (Mitterauer, 2005). But let us consider the
elementary pathophysiological mechanisms of schizophrenia as proposed. First of
all, there is a break of information processing between the glial system and
the neuronal system in tripartite synapses and also in the “orthogonal”
oligodendrocyte-axonic interaction. In this view, the term schizophrenia (split
of consciousness) is appropriate. In other words: a patient with schizophrenia
is permanently stressed by a world of intentions that cannot be mediated via
tripartite synapses to the neuronal system for reality testing. Such
considerations could be explanatory with concern to recent findings of
abnormalities in the white matter of the brain (Markis et al., 2010). Supposing
that a patient is under permanent pressure to realize his/her intentional
programs generated in the panglial syncytium, then the normal apoptosis could
be accelerated or mutations in astrocytes and in the oligodendrocyte-myelin
system could be activated. The effect is a decrease in white matter.
Considering the loss of oligodendrocytes which are normally interconnected with
astrocytes via gap junctions, the decay of oligodendrocytes must also destruct
the panglial syncytium in the sense of an increasing loss of gap junctions.
This loss of gap junctions may again destruct the capacity of the panglial
syncytium to generate intentional programs.
Many
patients with schizophrenia become increasingly psychobiologically exhausted in
the chronic course of their illness, which is called schizophrenic residuum. Of
course, the frequently observed disorders in neuronal networks may also play a
role, but the destruction of the panglial syncytium leading to a
dysintentionality per se, may be basically responsible for the negative view of
life, as typically seen in the schizophrenic residuum. Most impressively, if
the prefrontal cortex is severely affected, these patients are incapable of
planning. Therefore, what they want is merely the satisfaction of simple
biological needs (eating, drinking, smoking, getting money to buy something,
etc.). One could also say that with the destruction of the panglial syncytium
all kinds of destiny are broken down as well.
How
could astrocytes react to this disaster? Reactive astrocytosis may be a
compensatory attempt. Reactive astrocytosis occurs prominently in response to
all forms of CNS injury or disease. Recent studies point to the role of
reactive astrocytes in helping to limit tissue degeneration and preserve
function after CNS injury (Sofroniew, 2005). So why not in schizophrenia? If
one interprets the degeneration of the panglial syncytium caused by stress as a
functional brain injury, then reactive astrocytosis may here exert the same
mechanism. However, in schizophrenia astrocytosis may not only react to
injuries of the neuronal system, but also attempt to generate a new astrocytic
syncytium in reaction to the degeneration of the panglial syncytium. In this
way the patient can generate intentional programs and keep a “touch of destiny”
alive. This conjecture is experimentally
testable, if one compares the degree of dysintentionality – defined as the
ability to produce intentions or plans – of schizophrenic patients with and
without reactive astrocytosis.
Finally,
reactive astrocytosis can be seen in the light of the Astrocentric Hypothesis
(Robertson, 2002). According to this hypothesis, astrocytes represent the core
cells in the brain that not only control the glial-neuronal interaction, but
also determine the functions within the panglial syncytium. Therefore,
astrocytes may be capable – at least – to attempt repairing dysfunctions in the
panglial syncytium, as may be the case in schizophrenia. Admittedly, the pertinent
findings in brains with schizophrenia are contradictory. However, astrocytosis
(astrogliosis) may not occur widespread in the brain (Bernstein et al., 2008),
but it could represent a local phenomenon.
Pathophysiology
of non-schizophrenic delusions
The
pathophysiological mechanism of non-schizophrenic delusions may significantly
differ from schizophrenic delusions. Let us focus on the extrasynaptic fluid,
especially Ca2+ ions, in information transmission. Astrocytes sense
a decrease of Ca2+ in the extrasynaptic space via Cx43 hemichannels
and produce ATP that activates inhibitory interneurons, which negatively feed
back to the presynapse. I hypothesize that if Ca2+ is totally
exhausted (e.g. by stress), the negative feedback mechanism cannot be generated.
The exhaustion of Ca2+ may be caused by hyperexcitable neurons
flooding the synaptic neurotransmission. Importantly, the astrocytic receptors
are normally expressed, but they cannot cope with the synaptic neurotransmitter
flooding without an extrasynaptically generated negative feedback. Thus, in
non-schizophrenic delusions an unconstrained information flux may also occur,
but astrocytes per se may not be affected. Here, the brain operates in a
holistic and not schizophrenic way. This pathophysiological mechanism could
explain why patients with paranoid psychoses can mostly be successfully treated
with antipsychotic drugs. Their blockade of postsynaptic receptors constrains
neurotransmission such that astrocytes can work normally again. Although the neurons
may remain hyperexcited, the Ca2+ depletion is sufficient to
activate the negative feedback mechanism described. In parallel, the astrocytic
receptors are again able to modulate synaptic information processing, since
they can cope with the amount of neurotransmitters and temporarily turn off
synaptic information processing based on a negative feedback mechanism.
Do
glia play a comparable role in dream states and schizophrenic delusions?
One of
my schizophrenic patients spontaneously told me: “schizophrenia is dreaming,
that’s all”. Here, I will shortly attempt a “gliocentric” explanation that this
patient could be right. Despite progress in biological sleep or dream research,
up to now it is based on an exclusively neuronal approach (Hobson, 2005). However,
the current hypotheses on glial-neuronal interactions (Robertson, 2002;
Mitterauer, 2007; Mitterauer and Kopp, 2003) could be explanatory what the
alteration of consciousness in dreams concerns. Let me focus on strange dream
contents and scenarios. Strange dreams are defined as dream contents that are
unfeasible in wake conscious states. These are often compared to the main
schizophrenic symptoms of delusions and hallucinations. I hypothesize that the
same mechanism may be at work in the generation of both strange dreams and
schizophrenic delusions. It is experimentally verified that in tripartite
synapses astrocytes produce transmitters that occupy receptors on the
presynapse temporarily interrupting synaptic information transmission in the
sense of a negative feedback. In other words: astrocytes have a temporal
boundary-setting function (Mitterauer, 1998). The elementary mechanism may
exert an information structuring function determining the compartmental
organization of neuronal networks.
In
schizophrenia, this astrocytic information structuring function may be lost,
since astrocytes do not produce transmitters or express non-functional
receptors because of pertinent mutations. The effect is an unconstrained
information flux in synapses leading to compartmentless neuronal networks in
the sense of a generalization of neuronal information processing. I call this
“loss of self-boundaries in schizophrenia” (Mitterauer, 2003). Therefore, a
patient with schizophrenia is incapable of testing the reality of his (her)
delusional ideas. The phenomenological difference is that the dreaming person
upon awakening is fully able to test the reality. In contrast, the
schizophrenic patient is not. Supposing that our intentional programs are
permanently generated in the glial syncytia (Mitterauer, 2007), then their
realization in the neuronal networks via perception and motion is decisive. In
dreams we have the chance to play our intentions in various scenarios
independent of their feasibility, since the perception systems are turned off.
Now,
it is typical for strange dream scenarios that objects or individuals of the
environmental realities confuse and design uncanny figures and pictures in the
sense of a loss of boundaries. The same glial mechanism could be responsible as
in schizophrenic delusions. However, whereas schizophrenia represents a chronic
pathological process (mutations), dream states occur as a circadian
physiological behavior that gives us the chance of acting out our hidden
intentions without the pressure of their feasibility. Based on these
considerations the elementary function of glia in dream states could be
described as follows: astrocytes temporarily (according to the hypothalamic
circadian rhythms) turn off their interactions with the neuronal system in
tripartite synapses. This mechanism is comparable to that proposed for
schizophrenia. In addition, the generation of intentional programs in the glial
syncytia not only continues, but even dominates the consciousness as dreams
states.
Conclusion
My model
of impaired glial-neuronal interactions in schizophrenia is based on the core
hypothesis that nonfunctional astrocytic receptors may cause an unconstrained
synaptic information flux such that glia lose their modulatory function in
tripartite synapses. This may lead to a generalization of information
processing in the neuronal networks responsible for delusions and
hallucinations on the behavioral level. In this acute paranoid stage of
schizophrenia, nonfunctional astrocytic receptors or their loss decompose the
astrocyte domain organization with the effect that a gap between the neuronal
and the glial networks arises. If the illness progresses, the permanent
synaptic neurotransmitter flux may additionally impair the
oligodendrocyte-axonic interactions, accompanied by a “creeping” decay of
oligodendroglia, axons, and glial gap junctions responsible for severe
cognitive impairments. Here we may deal with aftereffects caused by the basic
fault of information processing in tripartite synapses. Importantly, the same
may hold true what the neuroinflammatory hypothesis (Brown, 2008) concerns. The
activation of microglia observed in brains with schizophrenia could represent a
reaction to the decay of nervous tissue described and not a primary
pathophysiological mechanism of schizophrenia.
Currently,
post mortem brains with schizophrenia are investigated with Storm microscopy, a
method that achieves resolution below the optical diffraction limit (University
of Applied Sciences, Linz, Austria). Whereas in the prefrontal cortex of normal
brains big amounts of receptors located on the processes of astrocytes are
found, in brains with schizophrenia astrocytic receptors cannot be identified
in this region. Since these preliminary results concern only serotonergic receptors
in the prefrontal cortex, further investigations of other receptor types on
astrocytes in pertinent brain regions is necessary. Should it be possible to
verify the central role of astrocytic receptors in the pathophysiology of
schizophrenia, then a diagnostic marker would be available, if an in vivo
identification is possible as well.
Acknowledgements
This
paper is dedicated to my friend and great neuroscientist Gerhard Werner (†),
University of Austin, Texas. I am also grateful to Birgitta Kofler-Westergren
for preparing the final version of this paper.
References
Alberdi, E.,
Sanchez-Gomez, M.V., Torre, I., Domercq, M., Perez-Samatin, A.,
Pérez-Cerdá, F., Matute, C., 2006.
Activation of kainate receptors sensitizes
oligodendrocytes to complement attack.
J Neurosci. 26, 3220-3228.
American
Psychiatric Association, 1998. Diagnostic and statistical manual of
mental disorders. American Psychiatric
Association, Washington.
Arajärvi, R.,
Varilo, T., Haukka, J., Suvissari, J., Suokas, J., Juvonen, H., Muhonen, M.,
Lönnquist, J., 2006. Affective
flattening and alogia associate
with familial form of schizophrenia.
Psychiatry Res. 141, 161-172.
Araque, A.,
Parpura, V., Sanzgiri, R.P., Haydon, P.G., 1999. Tripartite synapses:
glia, the unacknowledged partner.
Trends Neurosci. 22, 208-215.
Auld, D.S.,
Robitaille, R., 2003. Glial cells and neurotransmission: an inclusive
view of synaptic function. Neuron 40,
389-400.
Berge, S.,
Koenig, T., 2008. „Cerebral disconnectivity: an early event in schizophrenia.”
Neuroscientist 14, 19-45.
Bernstein, H.G., Steiner, J.,
Bogerts, B., 2009. „Glial cells in schizophrenia:
pathophysiological significance and
possible consequences for therapy.” Expert Rev.
Neurother. 9, 1059-1071.
Brown, A.S.,
2008. The risk for schizophrenia from childhood and adulthood infections.
Am. J. Psychiatry 165, 7-10.
Carpenter, W.T.,
Buchanan, R.W., 1995. Schizophrenia: introduction and overview,
In: Kaplan, H.J., Sadock, B.J. (Eds.),
Comprehensive Textbook of Psychiatry,
Williams and Wilkins, Baltimore, pp.
899-902.
De-Miguel, F.F.,
Fuxe, K., 2012. Extrasynaptic neurotransmission as a way of modulating
neuronal functions. Front. Physiol.
doi:10.3389/fphys2012.00016.
Deng, Z., Sobell,
J.L., Knowles, J.A., 2010. Epigenetic alterations in schizophrenia.
Focus 8, 358-365.
Dermietzel, R.,
Spray, D.C., 1998. From neuroglue to glia: a prologue. Glia 24, 1-7.
Di, X., Chan,
R.C., Gong, Q., 2009. White matter reduction in patients with
schizophrenia as revealed by voxel-based
morphometry: an activation likelihood
estimation meta-analysis. Prog.
Neuropsychopharmacol. Biol.
Psychiatry 33, 1390-94.
Erdi, P.,
Flaugher, B., Jones, T., Ujfalussy, B., Zalanyi, L., Diwadkar, V.A., 2007.
“Computational approach to
schizophrenia. Disconnection syndrome and dynamical
pharmacology.” AIP Conference
Proceedings 1028, 65-87.
Faa, V., Cobana,
A., Incani, F., Constantino, L., Cao. A., Rosatelli, M.C., 2010.
A synchronous mutation in the CFTR gene
causes aberrant splicing in an italian
patient affected by a mild form of
cystic fibrosis. J. Mol. Diagnostics 12, 380-383.
Fields,
R.D., 2009. The other brain, Simon and Schuster, New York.
Fields,
R.D., 2010. Change in the brain’s white matter. Science 330, 768-769.
Fields, R.D., Ni,
Y., 2010. Nonsynaptic communication through ATP release
from volume-activated anion channels in
axons. Sci. Signal. 3, ra 73.
Fisher, S.,
Cleveland, S.E., 1968. Body image and personality. Dover, New York.
Friston, K.J.,
1998. “The disconnection hypothesis. Schiz. Res. 30, 115-125.
Frith, C.D.,
1992. The cognitive neuropsychology of schizophrenia. Psychology
Press, East Sussex, UK.
Halassa, M.M.,
Fellin, T., Haydon, P.G., 2009. Tripartite synapses: roles for
astrocytic purins in the control of
synaptic physiology and behavior.
Neuropharm. 57, 343-346.
Halassa, M.M.,
Haydon, P.G., 2010. Integrated Brain Circuits: Astrocytic
Networks Modulate Neuronal Activity and
Behavior, Ann. Rev. Physiol.
72, 335-355.
Hashimoto, K., Engberg, G.,
Shimizu, E., Nordin, C., Lindstrom, L.H., Iyo, M.,
2005. Elevated glutamine/glutamate ratio in
cerebrospinal fluid of first episode
and drug naive schizophrenic patients.
BMC Psychiatry 5, 6.
Hobson, J.A.,
2005. Sleep is of the brain, by the brain and for the brain. Nature 437,
1254-1256.
Höstad, M.N,
Segal, D., Takahashi, N., et al., 2009. Linking white and grey matter
in schizophrenia: oligodendrocyte and
neuron pathology in the prefrontal
cortex. Front. Neuroanat. 3:9.
doi;10.3389/neuro.05.009.2009.
Ishibashi, T.,
Dakin, K.A., Stevens, B., Lee, P.R., Kozlov, S.V., Stewart, C.L.,
Fields, R.D., 2006. Astrocytes promote
myelination in response to
electrical impulses. Neuron 49,
823-832.
Kapur, S.,
Lecrubier, Y., 2003. Dopamine in the pathophysiology and treatment
of schizophrenia. Martin Dunitz,
London.
Káradóttir, R.,
Attwell, D., 2007. Neurotransmitter receptors in the life and death
of oligodendrocytes. Neuroscience 145,
1426-38.
Kettenmann, H.,
Ransom, B.R., 2005. Neuroglia. Oxford University Press, Oxford.
Kettenmann, H. and
Steinhäuser, C. (2005). „Receptors for neurotransmitters and
Hormones”, in: Kettenmann, H., Ransom,
B.R. (Eds.), Neuroglia, Oxford
University Press, Oxford, pp. 131-145.
Kyriakopoulos,
M., Perez-Iglesias, R., Woolley, J.B., Kamaan, R.A., Vyas, N.S.,
Barker, G.J., Frangou, S., McGuire,
P.K., 2009. Effect of age at onset
of schizophrenia on white matter
abnormalities. Br.
J. Psychiatry 195, 346-353.
Lenzenweger, M.F., McLachlan,
G., Rubin, D.B., 2007. Resolving the latent structure
of schizophrenia endophenotypes using
expectation-maximization-based finite
mixture modeling. J. Abn. Psychology 116, 16-29.
Markis, N., Seidman, L.J.,
Ahern, T., Kennedy, D.N., Caviness, V.S., Tsuang, M.T.,
Goldstein, J.M., 2010. White matter volume
abnormalities and association
with symptomatology in schizophrenia.
Psychiatry Res. 183, 21-29.
Meltzer, H.,
2003. „Multiple neurotransmitters involved in antipsychotic drug action”,
In: Kapur, S., Lecrubier, Y. (Eds.),
Dopamine in the pathophysiology and treatment
of schizophrenia, Martin Dunitz,
London, pp. 177-205.
Mitterauer, B.,
1998. An interdisciplinary approach towards a theory of
consciousness. BioSystems 45, 99-121.
Mitterauer, B.,
2003. The loss of self-boundaries: towards a neuromolecular theory
of schizophrenia. BioSystems 72,
209-215.
Mitterauer, B.,
2005. Nonfunctional glial proteins in tripartite synapses:
a pathophysiological model of
schizophrenia. Neuroscientist 11,
192-198.
Mitterauer, B.,
2007. Where and how could intentional programs be generated in
the brain? A hypothetical model based on glial-neuronal
interactions.
BioSystems 88, 101-112.
Mitterauer,
B., 2009. Loss of function of glial gap junctions may cause severe
cognitive impairments in schizophrenia.
Med. Hypotheses 73, 393-397.
Mitterauer, B.,
2010. Significance of the astrocyte domain organization for
qualitative information structuring in
the brain. Adv.
Biosci. Biotechnol. 1, 391-397.
Mitterauer, B., 2011. Qualitative
information processing in tripartite synapses: a
hypothetical model. Cogn. Comput. DOI
10.1007/s12559-011-9115-2.
Mitterauer, B.,
Kofler-Westergren, B., 2011. Possible effects of synaptic imbalances
on oligodendrocyte-axonic interactions
in schizophrenia: a hypothetical model.
Front. Psychiatry 2, doi:
10.3389/fpsyt.2011.00015.
Mitterauer, B., Kopp, C., 2003.
The self-composing brain: towards a glial-neuronal
brain theory. Brain and Cognition 51,
357-367.
Mitterauer, B., Pritz, W.F.,
1978. The
concept of the self: a theory of
self-observation. Int. Rev. Psychoanal.
5, 179-188.
Newman, E.A.,
2005. “Glia and Synaptic Transmission”, in: Kettenmann, H.,
Ransom, B.R. (Eds.), Neuroglia, Oxford University Press, Oxford, pp.
355-366.
Oberheim, N.A., Wang, X.,
Goldman, S., Nedergaard, M., 2006. Astrocytic
complexity distinguishes the human
brain, Trends Neurosci. 29, 547-553.
Pereira, A.,
Furlan, F.A., 2010. Astrocytes and human cognition: modeling
information integration and modulation
of neuronal activity. Prog.
Neurobiol. 92, 405-420.
Quintana-Murci,
L., 2012. Gene losses in the human genome. Science 335, 806-807.
Ransom, B.R., Ye,
Z., 2005. Gap junctions and hemichannels, in: Kettenmann, H.,
Ransom, B.R. (Eds.), Neuroglia, Oxford
University Press, Oxford, pp. 177-189.
Robertson,
J.M., 2002.The Astrocentric Hypothesis: proposed role of astrocytes
in consciousness and memory formation.
J. Phys. (Paris) 96, 251-255.
Rusakov,
D.A., 2012. Depletion of extracellular Ca2+ prompts astroglia to
modulate
synaptic network activity. Sci. Signal.
5, pe 4.
Saakov, B.A.,
Khoruzhaya, T.A., Bardakhchyan, E.A., 1977. Ultrastructural
mechanisms of serotonin demyelination.
Bulleten Eksperimental noi Biologii I
Meditsiny 83, 606-610.
Santello,
M., Volterra, A., 2010. Astrocytes as aide-mémoires, Nature 463, 169-170.
Shastry, B.S.,
2002. Schizophrenia: a genetic perspective. Int. J. Mol. Med. 9, 207-212.
Sims, A., 1991.
An overview of the psychopathology of perception; first rank symptoms
as a localizing sign in schizophrenia,
Psychopathology 24, 369-374.
Skelly, L.R.,
Calhoun, V., Meda, S.A., et al., 2008. Diffusion tensor imaging
in schizophrenia: relationship to
symptoms. Schizophr.
Res. 98, 157-162.
Steffek, A.E., 2007. „The role of
astrocytes in the pathophysiology of schizophrenia.”
Dissertation, University of Michigan,
159 pages, 3276299.
Stellwagen, D., Malenka, R.C.,
2006. Synaptic
scaling mediated by glial
TNF-alpha. Nature 440, 1054-1059.
Stevens, B.,
Porta, S., Haak, L.L., Gallo, V., Fields, R.D., 2002. Adenosine, a
neuron-glial transmitter promoting
myelination in the CNS in response to
action potentials. Neuron 36, 855-868.
Stevens,
B., Tanner, S., Fields, R.D., 1998. Control of myelination by specific patterns
of neural impulses. J. Neurosci.18,
9303-11.
Takahashi, N.,
Sakurai, T., Davis, K.L., Buxbaum, J.D., 2010. Linking
oligodendrocyte and myelin dysfunction
to neurocircuitry abnormalities in
schizophrenia. Prog. Neurobiol. 93,
13-24.
Torres, A., Wang,
F., Xu, Q., Fujita, T., Dobrowolski, R., Willecke, K., Takano, T.,
Nedergaard, M., 2012. Extracellular Ca2+
acts as a mediator of
communication from neurons to glia.
Sci. Sign. 208 ra8.
Wang, G., Cooper,
T.A., 2007. Splicing in disease: disruption of the splicing code
and the decoding machinery. Nature Rev.
Gen. 8, 749-761.
Wolf, R.C., Höse, A., Frasch,
K., Walter, H.,Vasic, N., 2008. Volumetric
abnormalities with cognitive deficits
in patients with schizophrenia.
Eur. Psychiatry 23, 541-548.
[ BWW Society Home Page ]
© 2013 The Bibliotheque: World Wide Society