Science: Chemistry:
Morphology of Silica Supported MoO3 Catalysts
By Dr. Jin-Wook Ha
Department of Chemical Engineering, Soonchunhyang University, Asan, Korea
ABSTRACT
1.
INTRODUCTION
Supported molybdenum trioxide catalysts have a vast abundance of industrial uses. Continual effort is put into characterization of the types of Mo oxide species which exist over various supports, their stability, and the optimal preparation techniques to obtain them. Various morphology influencing factors have been proposed for the impregnation and calcination steps of preparation. For example, a strong adsorption technique[1] has been employed to ensure that the negatively charged ionic complexes of molybdenum are uniformly adsorbed onto a positively charged surface(in an aqueous phase at a pH below the isoelectric point of the oxide). Many other groups have cited the importance of the oxide isoelectric point and other electrochemical factors regarding impregnation, a positive surface charge being necessary to achieve good dispersion of the negative Mo complexes adsorbed from liquid solution[2-4]. On the other hand, a number of groups have reported that well dispersed morphologies can be produced simply by calcining mechanical mixtures of MoO3 with the oxide supports[5-8], implying that morphology arises from a thermodynamic solid-solid wetting process.
A series of comprehensive works performed with Raman and UV over a number of supports and supported oxides and with a wide array of preparation procedures, including all those listed above, have synthesized many of these concepts into a general theory[9]. First, it was postulated that the surface species of molybdenum present in the air exposed, hydrated catalyst is only a function of pH in the hydrated layer, which can be calculated from the point of zero charge(or “pzc,” which is equivalent to the isoelectric point in the absence of specific adsorption) of the oxide, adjusted for the surface composition of molybdenum oxide or other oxide. The types of surface species are independent of the preparation procedure and complex. Second, the amount of material deposited in well dispersed form depends on the number of reactive hydroxyl groups present. For example, large amounts of well dispersed species can form over alumina and titania, which have high hydroxyl group surface densities, but only small amounts can form on silica, which has a low hydroxyl density[9]. Presumably, over the same support, allyl and chloride complexes exhibit a higher dispersion because they interact with the support at low temperatures at which more hydroxyl groups are present, whereas with typical impregnations with ammonium salts this interaction occurs at a high calcination temperature at which the surface is substantially dehydroxylated. This general picture would appear to be a good yardstick with which to compare any new characterization results of supported metal oxide systems.
In preliminary X-ray diffraction studies and in controlled atmosphere electron microscopy studies[10] of the molybdena/silica system, however, very interesting and potentially advantageous behavior of the hexagonal phase was observed. Whereas the formation of the orthorhombic phase at a high calcination temperature was accompanied by sintering and crystallite growth, at a lower temperature the formation of the hexagonal phase from large aggregates of the ammonium molybdate precursor was followed by continued disintegration of the crystallites, producing a highly dispersed form of MoO3 at relatively very high surface loadings. The present work with silica supported MoO3 has centered on investigating and exploiting the behavior of the little studied hexagonal phase of MoO3.
2. EXPERIMENTAL
For high surface area catalysts, Degussa Aerosil(fumed) silica was used. The powder was 99.8% pure with traces of A12O3, Fe2O3 , TiO2 and HCI. Na and Ca impurities in fumed silica are typically on the order of 2 ppm. The silica had a surface area of 380±30 m2/gm. To increase its bulk density, it was washed with deionized water and vacuum dried overnight at 40℃ and then air dried at ll0℃ for 12 hr. Ammonium heptamolybdate(IV) tetrahydrate, (NH4)6Mo7O24 ·4H2O,(AHM), was purchased from Aldrich. High loading samples were prepared by physically mixing the desired amount of dry support and precursor, and then adding sufficient water to reach incipient wetness. This technique circumvents the problem at high loadings that the amount of water added to the SiO2 is insufficient to disso1ve all of the AHM. This is typically resolved by performing successive impregnations with a lower concentration solution, whereas a single step was employed here. Acid or base impregnations were conducted with concentrated nitric acid(2.5 N) or ammonium hydroxide(5 N) in place of water. The acid preparation follows a standard procedure to produce hexagonal MoO3[11, 12], MoO3 weight loadings varied through 35.3% MoO3.
The most common weight loading studied, 26.7%, corresponds to a surface loading of 4 Mo atoms/nm2. The slurries were thoroughly stirred and dried in air overnight, after they had been spread in a thin 1ayer onto a watch glass. When samples were placed in a small crucible at depths of 1-2 cm, the top 2-3 mm of the sample turned darker(green) than the bulk of the sample after drying and calcination. Presumably some of the Mo containing phase migrated toward the surface during pretreatment. The final distribution of material appeared homogeneous with the use of watchglasses.
Other samples were made by physically mixing hexagonal or orthorhombic MoO3 and silica to the desired weight loadings, adding water to incipient wetness, and drying and calcining as before. This latter type of sample will be referred to as “physically mixed” and will be denoted “MoO3 + SiO2, ” whereas the former type will be referred to as “impregnated” and will be denoted “MoO3/SiO2”.
For low loading, high surface area samples used in conjunction with XPS measurements, except that to the physical mixtures of support and precursor an excess of liquid was added(wet impregnation) so that pH could be controlled and measured. These mixtures were dried in a rotary vacuum drier at 40℃ and subsequently calcined in a muffle furnace. The spectroscopy was performed at the Amoco Research Center in Naperville, Illinois, using an SSX 100 spectrometer with an A1Kα source at 25℃. All samp1es contained Mo in the +6 oxidation state. The Mo/Si ratio was used as an estimate of dispersion.
Mode1, low surface area catalysts for electron microscopy were prepared from planar silica substrates and nonporous silica spheres as described[10]. Brief1y, dry impregnations as described above with a surface loading of 4 atoms Mo/nm2, were performed with the spherical silica powder. Samples were dispersed onto holey carbon substrates on gold finder grids. Catalyst impregnation was mimicked over the planar silica substrate by depositing a drop of the precursor solution onto the substrate and allowing the water to evaporate in air. Gold finder substrates were also used with the planar samples. A particular area of a sample was photographed through a series of calcination treatments microscopy was performed with a JEOL 100 CX, operated at 100 kV.
3. RESULTS AND DISCUSSION
Transmission Electron Microscopy(TEM) Characterization of Model Catalysts
An ammonium molybdate precursor, deposited on a planar substrate by allowing a drop of 2*10-3 molar solution to dry at room temperature on the substrated grid, is shown in Fig. 1A just after drying.
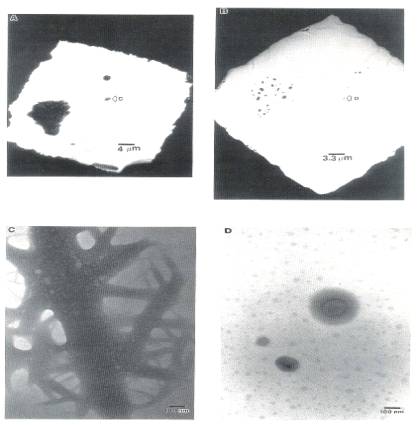
Fig. 1. TEM micrographs of calcined MoO3/SiO2:
(A) AHM impregnated, dried; (B) sample(A) calcined at 500℃, 2hr; (C) closeup region of dried sample; (D) closeup
of same region as (C) in calcined sample.
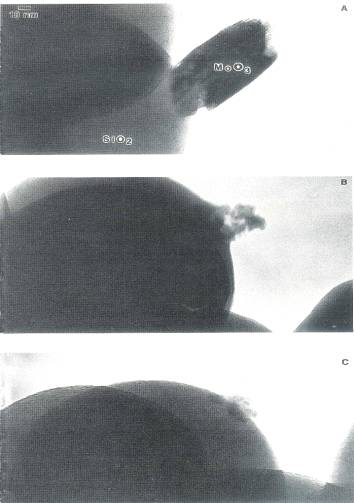
Fig. 2. TEM micrographs of calcined MoO3/non
porous spherical SiO2: (A) AHM impregnated, calcined 300℃, 2hr; (B) sample (A) calcined an additional 4 hr; (C)
sample (B) calcined at 500℃,
2hr.
This precursor was mainly in the form of very large bundled aggregates, with a number of maller such aggregates, one of which is labeled "C" in the figure. After a 500℃ calcination, the precursor had spread dramatically(Fig. 1B). A higher magnification image of region "C" from Fig. 1A is shown in Fig. 1C before calcination, and in Fig. 1D after calcination. The original material almost completely disintegrates, leaving only three smaller crystallites. These exhibit aprons of surrounding material. In addition, patches of material appeared over the substrate. These did not diffract, and neither was a third dimension apparent upon changing the sample orientation, meaning that the patches were thin. These 100 to 400Å patches are somewhat similar to small, ill defined MoO3, patches observed on alumina[13], although larger in size. This behavior implies that the material can drastically spread as the precursor transforms to the trioxide. This change in morphology could likely have occurred during the warmup to 500℃. Appreciable migration of material was observed in another TEM study as a supported Mo metal and Mo suboxide sample was transformed to trioxide by heating from 200 to 300℃ in air[11].
The dispersion of the hexagonal phase and its conversion to the orthorhombic is illustrated in Fig. 2. The very large crystallite attached to the silica sphere in Fig. 2A was produced by a 300℃, 2 hr air calcination. It was found to be hexagonal from an analysis of its spot electron diffraction pattern. Most of the area of the silica sphere was empty. After an additional 300℃, 4 hr calcination, shown in Fig. 2B, most of the MoO3, appeared to have dispersed over the silica sphere. This finding supports the conclusion that the hexagonal phase disperses over the silica surface. Figures 2A and 2B illustrate the further dispersion of the highest loading hexagonal MoO3. The remaining multigrained cluster in Fig. 2B appears to wet the silica surface. Upon a 500℃ calcination for 2 hr, the crystallite cluster's volume decreased somewhat further and individual grains in it appeared to have sintered together. The sintered crystallite would produce a sharper XRD peak, while the multigrained aggregate in Fig. 2B would produce a peak of similar integrated intensity, but of smaller height and greater breadth The area was not monitored after further calcination time to see whether the orthorhombic crystallite grew more in size. As another indication of a weaker MoO3/SiO2 interaction, however, a smooth junction of MoO3, with the silica surface is no longer apparent in Fig. 2C; it does not appear that this crystallite strongly wets the surface. In an earlier work[10] the stability of the orthorhombic phase at 500℃ was documented.
Transformation of the hexagonal crystallites, produced by another 300℃, 2 hr calcination of the dried precursor, Fig. 3A, into the orthorhombic form by a 500℃, 2 hr calcination is shown in Fig. 3B. The volume of the crystallite phase did increase considerably in this instance. In fact, some material appears to have come from the surrounding grid bar. Fig. 3 is indicative of the many XRD sequences in which the sintered orthorhombic phase was produced from the hexagonal phase. The same trends in dispersion with respect to calcination temperature, that is, increasing dispersion during a low temperature calcination of the hexagonal phase and decreasing dispersion during a high temperature calcination of the orthorhombic phase, were reported in an earlier work[10].
X-ray Photoelectron Spectroscopy(XPS) Characterization of Low Loading Catalysts
Precursor and calcined samples of 1.8 and 6.8 weight percent catalysts, or 0.2 and 0.8 Mo atom/nm2 surface loading, were characterized with XPS. For each loading, two initial pH values were used, one below the point of zero charge of silica, where strong adsorption of negatively charged molybdate ions can occur over a positively charged silica surface[1-4], and the other over a pH of 9, far above the pzc and so over a negatively charged silica surface. After the impregnation and drying at 40℃ for 2 hr in a rotary vacuum drier, samples were calcined at both 300 and 500℃ for 2 and 6 hr. Mo dispersion was estimated by the Mo/Si ratio; this is charted as a function of initial pH, calcination temperature, time, and loading in Fig. 4. For both loadings the initial dispersion of the high pH sample was much higher than the low pH sample: a factor of almost 2.5 higher for the 0.2 Mo atom/nm2 sample, and over 5 times higher for the 0.8 Mo atom/nm2 sample. In every case, the dispersion of the highly dispersed samples decreased upon calcination, while that of the poorly dispersed precursors increased upon calcination. Because the final dispersion is below maximum, the "patchy monolayer” morphology is evidenced in all of the calcined samples.

Fig. 3. TEM micrographs of calcined MoO3/non
porous spherical SiO2: (A) AHM impregnated, calcined 300℃, 2hr; (B) sample (A) calcined at 500℃, 2hr.
The final dispersion for the respective weight loadings appeared to be almost independent of initial dispersion, and only a weak function of calcination temperature, For the high loading catalyst, the Mo/Si ratio of the 500℃ calcined samples is about 20% higher than the average final Mo/Si ratio of the 300℃ calcined sample, 32*10-3 to 25*10-3 . The average final Mo/Si ratios of the lowest loading samples are about the same, 8*10-3. The final surface compositions are roughly proportional to loading, These results support the earlier TEM experiment(Fig. 1) which shows that drastic rearrangement of material can occur in the conversion from precursor to trioxide.
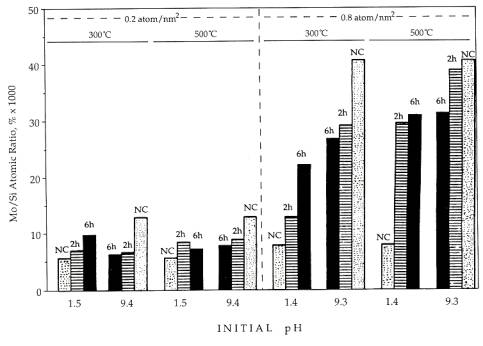
Fig.
4. XPS Mo/Si ratios as functions of impregnation pH, Mo loading, and calcinations
temperatures.
CONCLUSIONS
All morphologies of silica supported MoO3 appear to be thermodynamically driven. At low loadings there appears to be one stable morphology and dispersion, which agrees with recent in situ Raman data. For high loaded calcined catalysts there appear three states: a metastable sintered hexagonal state and a well dispersed hexagonal state at moderate temperatures, and a sintered orthorhombic state at high temperatures(≤ 400-500℃). It is possible to produce the well dispersed hexagonal phase from the sintered orthorhombic phase with an ammonia impregnation, which forms the ammonium rich precursor phases, and subsequent calcination at 300℃. The apparent increase in dispersion of the hexagonal phase may arise from some role of ammonia which results in a stronger MoO3-SiO2, interaction.
REFERENCES:
1. Wang, L., and Hall, W. K., J. Catal., 77, 232(1982).
2. Okamote, Y., and Imanaka, T., J. Phys. Chem., 92, 7102(1988).
3. CaCeres, C. V., Fierro, J. L. G., Lazaro, J., Lopez, A., and Soria, J., J. Catal., 122, 113(1990).
4. Spanos, N., Vordonis, L., Koutsoukos, P. G., and Lycourghiotis, A., J. Catal., 124, 301(1990).
5. LeYer, J., Zaki, M. I., and Knozinger, H., J. Phys. Chem., 90, 4775(1986).
6. Stampfl, S. R., Chen, Y., Dumesic, J. A., Niu, C., and Hil1 Jr., C. G., J. Catal., 105, 445(1987).
7. Haber, J., Machez, T., and Grabowski, R., Solid State Ionic J., 32/33, 887(1989).
8. Leyer, J., Mey, D., and Knozinger, H., Surf. Sci., 201, 603(1990).
9. Wachs, I. E., Chem. Eng. Sci., 45, 2561(1990).
10. Datta, A., Ha, J. W., Regalbuto, J. R., and Alien, C. W., Ultramicroscopy, 29, 233(1989).
11. Greve, C. L. and Schmidt, L. D., Appl. Surf. Sci., 35, 199(1988-1989).
12. Noh, J. S. and Schwarz, J. A., J. Colloid Interface Sci., 130, 157(1989).
13. Hayden, T. F. and Dumesic, J. A., J. Catal., 103, 366(1987).
[ BWW Society Home Page ]
© 2003 The BWW Society/The Institute for the Advancement of Positive Global Solutions