Science: Chemistry:
The Equilibrium Model of MoO3 Containing
Phases Supported in Silica
Jee Nyu Hong and Dr. Jin-Wook Ha
Department of Chemical Engineering, Soonchunhyang University
Editor's note: BWW Society member Dr. Jin-Wook Ha conducts his research in the Department of Chemical Engineering at Korea's Soonchunhyang University; for another research paper published in this journal by Dr. Ha, refer to Characterization of Polyaniline Synthesized in Presence of Azo-initiator (VA-044).
ABSTRACT
The
morphology of silica supported MoO3 catalysts, which was prepared by
impregnation of ammonium heptamolybdate with various weight loadings up to 35
wt%, was studied using x-ray diffraction. In addition to the orthorhombic
phase, the behavior of the rarely studied hexagonal phase was characterized.
For high 1oading catalysts, excess ammonium ions present in the monoclinic and
triclinic precursors are capable of occupying interstitial sites of
microcrystalline MoO3 during moderate temperature calcinations and
in doing so enhance the MoO3-SiO2 interaction. This
results in a "well dispersed" morphology at high loadings. Sintering
at high temperature is due to loss of ammonium from the oxide framework.
Ammonia reimpregnation, which leads back to the well dispersed hexagonal phase,
may offer a simple regeneration process for spent Mo containing catalysts.
Key
Words: Equilibrium Model, Silica Supported MoO3 Catalyst
1. INTRODUCTION
Supported
molybdenum trioxide catalysts have a vast abundance of industrial uses.
Continual effort is put into characterization of the types of Mo oxide species
which exist over various supports, their stability, and the optimal preparation
techniques to obtain them. Various morphology influencing factors have been
proposed for the impregnation and calcination steps of preparation [1-5].
However, there is still dispute for the nature of surface species over silica.
Many reports suggest that the monomeric surface species exist at low loadings,
polymeric at intermediate loadings, and bulk at high loadings, roughly in
parallel to alumina, except that the formation of the polymolybdate and the
bulk species occurs at lower loadings. More recent in situ works [6], in
which only the polymeric species is observed before the formation of the bulk
form at higher loadings, have brought this idea into question. According to the
general synthesis theory as pertains silica [7], the pzc of silica is very low
and so low pH values of the hydrated layer are always present giving rise only
to the polymeric, octahedral species Tetragonal signals were shown to arise
from NaMoO3 and CaMoO4 crystalline impurities stemming
from Na and Ca impurities in precipitated silica. The present work with silica
supported MoO3 has focused on investigating and exploiting the
behavior of little studied hexagonal phase of MoO3. From
experimental results, equilibrium model of MoO3 containing phases
supported in silica was suggested.
2. EXPERIMENTAL
For
high surface area catalysts, Degussa Aerosil (fumed) silica which had a surface
area of 380 m2/g was used. To increase its bulk density, it was
washed with deionized water and vacuum dried overnight at 40 and then air dried at 110C for 12h. Ammonium heptamolybdate (IV)
tetrahydrate (AHM), (NH4)6Mo7O24•4H2O,
was purchased from Aldrich. High loading samples were prepared by physically
mixing the desired amount of dry support and precursor, and then adding
sufficient water to reach incipient wetness. Acid or base impregnations were
conducted with concentrated nitric acid (2.5N) or ammonium hydroxide (5N)
in place of water. The acid preparation follows a standard procedure to produce
hexagonal MoO3 [8]. MoO3 weight loadings varied through
35.3 wt% MoO3. The most common weight loading studied, 26.7 wt%,
corresponds to a surface loading of 4 Mo atoms/nm2. The slurries
were thoroughly stirred and dried in air overnight, after they had been spread
in a thin 1ayer onto a watch glass. Calcinations were performed at either 300
or 500C in air. Unless otherwise specified,
calcination times were 2h. Other samples were made by physically mixing
hexagonal or orthorhombic MoO3 and silica to the desired weight
loadings, adding water to incipient wetness, and drying and calcining as
before. This latter type of sample will be referred to as physically mixed and
will be denoted MoO3+SiO2, whereas the former type will
be referred to as impregnated and will be denoted MoO3/SiO2.
All samples were characterized by x-ray diffraction(XRD).
3. RESULTS AND DISCUSSION
X-ray
diffraction patterns for the unsupported and supported precursors calcined at
300 or 500C are shown in Figure 1. All of the three
different unsupported precursor phases formed orthorhombic (denoted “O” in the
Figures) form of MoO3 after a 500C calcination. Only the pattern which
resulted from the neutral pH precursor, (NH4)6Mo7O24•4H2O,
is shown [pattern (g)]; the others are identical. Additionally, the unsupported
neutral pH and the basic precursors largely formed the orthorhombic phase upon
a 300C calcinations [patterns (f) and (e)],
though a small amount of the hexagona1 (denoted “H” in the Figures) phase was
also seen for the basic precursor.
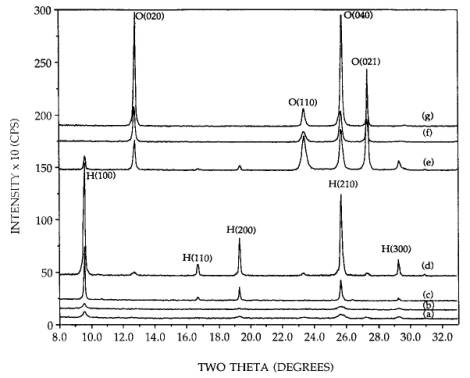
Figure 1. XRD patterns of calcined 36.5 wt% supported
and unsupported MoO3: (a) 300C calcined supported basic precursor (hexagonal), (b) 300C calcined supported neutral precursor (hexagonal), (c) 300C calcined supported acid precursor (hexagonal), (d) 300C calcined unsupported acid precursor (hexagonal), (e) 300C calcined unsupported basic precursor (orthorhombic and some
hexagonal), (f) 300C calcined unsupported neutral precursor (orthorhombic), (g) 500C calcined supported neutral precursor (orthorhombic).
The
peak breadths of the orthorhombic phases produced at 300C are somewhat broader than those formed
at 500C meaning that they are more poorly
crystallized. The unsupported and supported hexagonal precursor phases
[patterns (d) and (c)] maintained the hexagonal phase after the calcination,
but sintered somewhat as the peak breadths have reduced. Finally, the 300C calcination of the neutral and basic
precursor phases resulted in a relative1y very dispersed hexagonal phase. The
crystallite size of the fraction of crystallites observable by XRD was
estimated from the Scherrer equation to be 320 Å, using the full width at half
maximum of the unsupported phases as standards for instrumental broadening.
However, since the integrated intensities of the dispersed phase are well below
those of the sintered phases, a good portion of the material also exists as
very small (< 40 Å) particles beyond the detection limit of XRD. These could
be truly amorphous or microcrystalline. No material was lost through
volatilization; a high degree of crystallinity can be reestablished in the
dispersed samples by subsequent high temperature treatments as will be shown in
later figures. Samples in which AHM powder was not thoroughly mixed with SiO2
before or after the addition of water yielded the orthorhombic phase and not
the well dispersed hexagonal after a 300C calcination. Proximity to the silica
surface is thus necessary for the hexagonal phase to be produced.
The
interconversion between the hexagonal and orthorhombic phases for 26.7 wt%
samples, is illustrated in Figure 2, starting with the dispersed hexagonal
phase [pattern (a)]. After an additional 4h of calcination at 300C [pattern (b)], the small hexagonal peaks
had diminished completely. In some trials, however, the small peaks observed
after 2h [pattern (a)] remained after 6h calcination time. This may again be an
effect of the thoroughness of mixing of he sample. The pattern of the
orthorhombic phase which forms upon a direct, 500C calcination for 2h is shown in pattern
(c); an additional 4h calcination at 500C heightens the peaks by about 5 or 10%.
The well dispersed hexagonal phase is converted to the sintered orthorhombic
upon calcination at 500C [pattern (d)], and again continued
calcination for 4h at 500C increases the peak heights only
slightly. Pattern (e), which exhibits the same sintered orthorhombic pattern
and intensity as patterns (c) and (d), was taken after a 300C, 6h calcination of the sintered orthorhombic
phase [pattern (c)] and shows that the hexagonal-orthorhombic phase transition
for the supported phases is irreversible in air, as reported for unsupported
compounds [9, 10]. The same set of calcinations were performed with the basic
(monoclinic plus triclinic) precursor, with virtually the same results. That
is, the crystalline phases, peak breadths, and intensities produced by the
neutral and basic precursors are essentially the same upon calcination at or
above 300C.
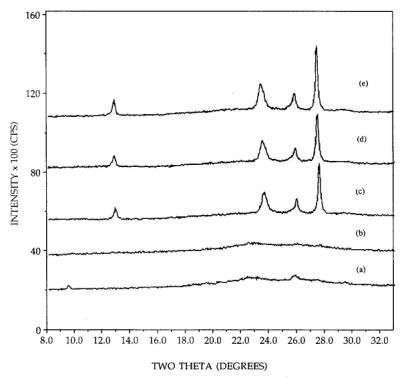
Figure 2. XRD patterns of 26.7 wt% MoO3,
calcinations of: (a) 300C, 2h, (b) 300C, 6h, (c) 500C, 2h, (d) 300C, 6h and 500C, 2h, (e) 500C, 2h and 300C, 6h.
The
fraction of MoO3 in the hexagonal and orthorhombic phase observable
by XRD was found from the experiment illustrated in Figure 3. Pure hexagonal
MoO3 was mixed to 26.7 wt% with SiO2 powder [pattern
(a)]. The acid impregnated and 300C calcined hexagonal sample is shown in
pattern (b). While the peaks heights are somewhat lower, they are also broader;
the integrated are as are equal, meaning that virtually all the sintered
hexagonal phase is observed by XRD. In contrast, almost all of the well
dispersed hexagonal phase [pattern (c)] is below the limit of detection of XRD.
A 26.7 wt% physical mixture of orthorhombic MoO3+SiO2 is
shown in pattern (d), and a 26.7 wt% impregnated orthorhombic sample in pattern
(e). The integrated intensities of the impregnated sample are about 90-95% of
those of the physically mixed sample, meaning that the vast majority of MoO3
is observable by XRD. Thus XRD provides a good measure of the difference in
dispersion of the two phases, and this difference is quite large.
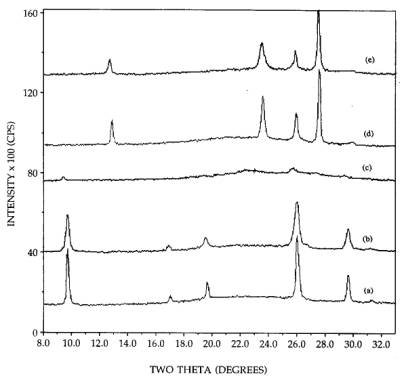
Figure 3. XRD patterns of 26.7 wt% MoO3:
(a) hexagonal MoO3+SiO2, (b) hexagonal MoO3/SiO2,
300C calcinations (acid impregnation), (c) hexagonal MoO3/SiO2,
300C calcinations (neutral impregnation), (d) orthorhombic MoO3+SiO2,
(e) orthorhombic MoO3/SiO2.
The
dramatic effect of ammonia on the dispersion of silica supported MoO3
was found in a series of experiments with physical mixtures of silica and
hexagonal MoO3 (formed by the acid precursor and 300 calcination) and orthorhombic MoO3.
The first experiment is shown in Figure 4A, for the physically mixed hexagonal
MoO3+SiO2. Pattern (a) was taken from the initial
mixture. On continued calcination at 300C [patterns (b) and (c)] the morphology
remained stable. However, when the initial sample was reimpregnated to
incipient wetness with a solution containing 5N NH4OH, dried
at room temperature, and calcined 2h at 300C, drastic spreading occurred pattern
(d)]. The behavior of a physically mixed silica and orthorhombic sample is
shown in Figure 4B. The orthorhombic MoO3 for these samples was
prepared by an acid impregnation of AHM and calcination at 500C. The acid was employed to make the
preparation conditions otherwise identical to the physically mixed hexagonal
sample. Similar to the hexagona1 MoO3+SiO2 physical
mixture, the pattern of orthorhombic MoO3 was unchanged after an air
calcination at 300C for 2h [pattern (b)] and was slightly
diminished in intensity after 4h calcination time [pattern (c)].
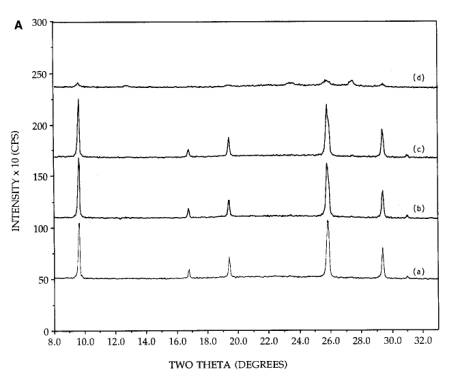
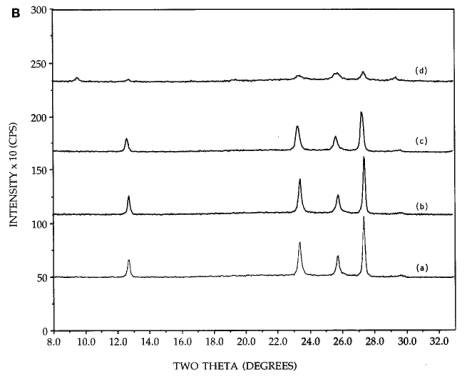
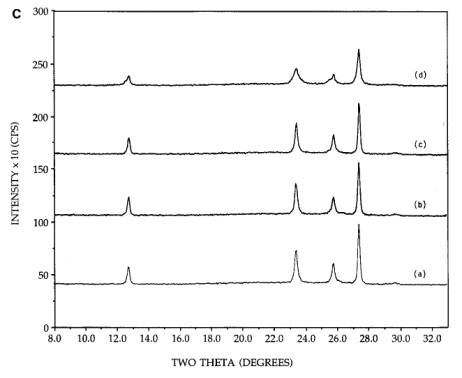
Figure 4. [A] XRD patterns of 26.7 wt%
physically mixed hexagonal MoO3+SiO2: (a) initial
mixture, (b) after 300C, 2h calcination, (c) after 300C, 4h calcination, (d) after reimpregnation with ammonia solution,
calcination for 300C, 2h. [B] XRD patterns of 26.7 wt% physically mixed
orthorhombic MoO3+SiO2: (a) initial mixture, (b) after
300C, 2h calcination, (c) after 300C, 4h calcination, (d) after reimpregnation with ammonia solution,
calcination for 300C, 2h. [C] XRD patterns of 26.7 wt% physically mixed
orthorhombic MoO3+SiO2: (a) initial mixture, (b) after
500C, 2h calcination, (c) after 500C, 4h calcination, (d) after reimpregnation with ammonia solution,
calcination for 500C, 2h.
After
reimpregnation of the initial sample with the 5N ammonia solution,
drying, and another 300C, 2h calcination as before, not only was
spreading again drastic, but the majority of the observable crystalline phase
was present in the hexagonal form [pattern (d)]. The final degree of
crystallinity of both samples, Figures 4A and 4B, approaches that of the
dispersed, neutral and basic impregnated precursors (Figures 1-3). While there
are small amounts of orthorhombic remaining, this morphology will also be
referred to as the well dispersed hexagonal phase. In Figure 4C, the physically
mixed orthorhombic and silica sample [pattern (a)] shows little or no change in
morphology upon continued calcination at 500C [patterns (b) and (c)], in agreement
with similar experiments [11, 12]. After impregnation with ammonia solution and
a 500, 2h calcination, some peak broadening
was observed [pattern (d)], but no conversion to the hexagonal phase was
evidenced.
The
reversibility of MoO3 wetting of silica via ammonia reimpregnation
and the hexagonal to orthorhombic phase change were examined in a further XRD
experiment. In Figure 5, the physically mixed orthorhombic MoO3+SiO2
mixture is again shown, pattern (a), and the redispersed morphology formed by
ammonia impregnation and a 300C, 2h calcination is shown in pattern (b).
A 500C, 2h calcination produces the sintered
orthorhombic phase once again with no hexagonal phase remaining, pattern (c),
while a second ammonia impregnation and 300C calcination produces the dispersed
mixture of phases again, pattern (d).
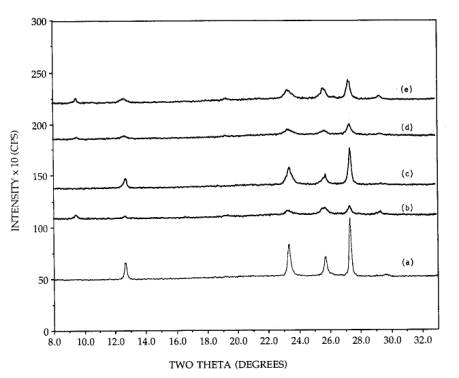
Figure 5. XRD patterns of 26.7 wt% supported MoO3:
(a) 500C calcined, orthorhombic MoO3/SiO2, (b)
ammonia reimpregnated and 300C calcined, (c) 500C calcined, (d) ammonia reimpregnated and 300C calcined, (e) acid impregnated sample, 300C calcined.
In
another experiment not shown, distil1ed water was used in place of an ammonia
solution in an attempt to redisperse orthorhombic MoO3 without
ammonia, however, no change occurred. Thus, the redispersion and sintering
processes are reversible contingent upon the presence of ammonia. In an attempt
to remove the ammonia from a well dispersed sample, a portion of this catalyst
was reimpregnated with nitric acid, and then calcined at 300C for 2h. The resulting pattern is shown
as pattern (e); some sintering had been induced by the acid treatment. The
pathway from the sintered hexagonal or orthorhombic phases to the well
dispersed hexagonal phase, as seen in Figures 4A and 4B, appears at least in
part to pass through the basic precursor phase. From there it becomes parallel
to decomposition of the impregnated basic precursor. From these experimental results,
equilibrium phase model of MoO3 containing phases supported in
silica was suggested and shown in Figure 6.
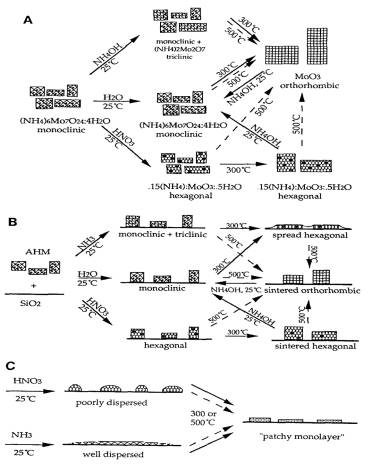
Figure 6. The equilibrium phase model of MoO3
containing phases supported in silica:
A. unsupported MoO3, B. high
loaded MoO3 over silica, C. low loaded MoO3 over silica.
In
summary, for high 1oading catalysts is that excess ammonium ions present in the
monoclinic and triclinic precursors are capable of occupying interstitial sites
of microcrystalline MoO3 during moderate temperature calcinations
and in doing so enhance the MoO3-SiO2 interaction. This
results in a "well dispersed" morphology at high loadings. Sintering
at high temperature is due to loss of ammonium from the oxide framework.
Ammonia reimpregnation, which leads back to the well dispersed hexagonal phase,
may offer a simple regeneration process for spent Mo containing
catalysts.
4. CONCLUSIONS
All
morphologies of silica supported MoO3 appear to be thermodynamically
driven. At low loadings there appears to be one stable morphology and
dispersion, which agrees with recent in situ XRD data [13]. For high loaded
calcined catalysts there appear three states: a metastable sintered hexagonal
state and a well dispersed hexagonal state at moderate temperatures, and a
sintered orthorhombic state at high temperatures (400-500C). It is possible to produce the well
dispersed hexagonal phase from the sintered orthorhombic phase with an ammonia
impregnation, which forms the ammonium rich precursor phases, and subsequent
calcination at 300C. The apparent increase in dispersion of
the hexagonal phase may arise from some role of ammonia which results in a
stronger MoO3-SiO2 interaction.
REFERENCES:
1. R.
Margraf, J. Leyrer, E. Taglauer, and H. Knotinger, React. Kinet. Catal.,
35, 261 (1987).
2. Y.
Okamote and T. Imanaka, J. Phys. Chem., 92, 7102 (1988).
3. C.
V. CaCeres, J. L. G. Fierro, J. Lazaro, A. Lopez, A. Agudo, and J. Soria, J.
Catal., 122, 113 (1990).
4. F.
M. Mulcahy, M. J. Fay, A. Proctor, M. Houalla, and D. M. Hercules, J.
Catal., 124, 231 (1990).
5. N.
Spanos, L. Vordonis, P. G. Koutsoukos, and A. Lycourghiotis, J. Catal.,
124, 301 (1990).
6. N.
Kakuta, K. Tohji, and Y. Udagawa, J. Phys. Chem., 92, 2583 (1988).
7. C.
P. Cheng and G. L. Schrader, J. Catal., 60, 276 (1979).
8. C.
L. Greve, and L. D. Schmidt, Appl. Suuf. Sci., 35, 199 (1989).
9. N.
A. Caiger, S. Crouch Baker, P. G. Dickens, and G. S. James, J. Solid State Chem.,
67, 369 (1987).
10. I.
P. Olenkova, D. V. Tarasova, G. N. Kustova, G. I. Aleshina, and E.
L. Mikhailenko, React. Kinet. Catal. Lett., 9, 221 (1978).
11. S.
R. Stampfl, Y. Chen, J. A. Dumesic, C. Niu, and Jr. C. G. Hill, J. Catal.,
105, 445 (1987).
12. J.
Leyer, D. Mey, and H. Knozinger, J. Catal., 124, 349 (1990).
13. J.-W.
Ha, J. of Ind. and Eng. Chem., 4(3), 191 (1998).
[ BWW Society Home Page ]
© 2002 The BWW Society/The Institute for the Advancement of Positive Global Solutions